Title 40
PART 136 APPENDIX A
| Parameter | STORET No. | CAS No. |
|---|---|---|
| Bromodichloromethane | 32101 | 75-27-4 |
| Bromoform | 32104 | 75-25-2 |
| Bromomethane | 34413 | 74-83-9 |
| Carbon tetrachloride | 32102 | 56-23-5 |
| Chlorobenzene | 34301 | 108-90-7 |
| Chloroethane | 34311 | 75-00-3 |
| 2-Chloroethylvinyl ether | 34576 | 100-75-8 |
| Chloroform | 32106 | 67-66-3 |
| Chloromethane | 34418 | 74-87-3 |
| Dibromochloromethane | 32105 | 124-48-1 |
| 1,2-Dichlorobenzene | 34536 | 95-50-1 |
| 1,3-Dichlorobenzene | 34566 | 541-73-1 |
| 1,4-Dichlorobenzene | 34571 | 106-46-7 |
| Dichlorodifluoromethane | 34668 | 75-71-8 |
| 1,1-Dichloroethane | 34496 | 75-34-3 |
| 1,2-Dichloroethane | 34531 | 107-06-2 |
| 1,1-Dichloroethane | 34501 | 75-35-4 |
| trans-1,2-Dichloroethene | 34546 | 156-60-5 |
| 1,2-Dichloropropane | 34541 | 78-87-5 |
| cis-1,3-Dichloropropene | 34704 | 10061-01-5 |
| trans-1,3-Dichloropropene | 34699 | 10061-02-6 |
| Methylene chloride | 34423 | 75-09-2 |
| 1,1,2,2-Tetrachloroethane | 34516 | 79-34-5 |
| Tetrachloroethene | 34475 | 127-18-4 |
| 1,1,1-Trichloroethane | 34506 | 71-55-6 |
| 1,1,2-Trichloroethane | 34511 | 79-00-5 |
| Tetrachloroethene | 39180 | 79-01-6 |
| Trichlorofluoromethane | 34488 | 75-69-4 |
| Vinyl chloride | 39715 | 75-01-4 |
1.2 This is a purge and trap gas chromatographic (GC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compounds above, compound identifications should be supported by at least one additional qualitative technique. This method describes analytical conditions for a second gas chromatographic column that can be used to confirm measurements made with the primary column. Method 624 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for most of the parameters listed above.
1.3 The method detection limit (MDL, defined in Section 12.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.4 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.5 This method is restricted to use by or under the supervision of analysts experienced in the operation of a purge and trap system and a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 An inert gas is bubbled through a 5-mL water sample contained in a specially-designed purging chamber at ambient temperature. The halocarbons are efficiently transferred from the aqueous phase to the vapor phase. The vapor is swept through a sorbent trap where the halocarbons are trapped. After purging is completed, the trap is heated and backflushed with the inert gas to desorb the halocarbons onto a gas chromatographic column. The gas chromatograph is temperature programmed to separate the halocarbons which are then detected with a halide-specific detector. 2 3
2.2 The method provides an optional gas chromatographic column that may be helpful in resolving the compounds of interest from interferences that may occur.
3. Interferences3.1 Impurities in the purge gas and organic compounds outgassing from the plumbing ahead of the trap account for the majority of contamination problems. The analytical system must be demonstrated to be free from contamination under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3. The use of non-Teflon plastic tubing, non-Teflon thread sealants, or flow controllers with rubber components in the purge and trap system should be avoided.
3.2 Samples can be contaminated by diffusion of volatile organics (particularly fluorocarbons and methylene chloride) through the septum seal ilto the sample during shipment and storage. A field reagent blank prepared from reagent water and carried through the sampling and handling protocol can serve as a check on such contamination.
3.3 Contamination by carry-over can occur whenever high level and low level samples are sequentially analyzed. To reduce carry-over, the purging device and sample syringe must be rinsed with reagent water between sample analyses. Whenever an unusually concentrated sample is encountered, it should be followed by an analysis of reagent water to check for cross contamination. For samples containing large amounts of water-soluble materials, suspended solids, high boiling compounds or high organohalide levels, it may be necessary to wash out the purging device with a detergent solution, rinse it with distilled water, and then dry it in a 105 °C oven between analyses. The trap and other parts of the system are also subject to contamination; therefore, frequent bakeout and purging of the entire system may be required.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 4 6 for the information of the analyst.
4.2 The following parameters covered by this method have been tentatively classified as known or suspected, human or mammalian carcinogens: carbon tetrachloride, chloroform, 1,4-dichlorobenzene, and vinyl chloride. Primary standards of these toxic compounds should be prepared in a hood. A NIOSH/MESA approved toxic gas respirator should be worn when the analyst handles high concentrations of these toxic compounds.
5. Apparatus and Materials5.1 Sampling equipment, for discrete sampling.
5.1.1 Vial - 25-mL capacity or larger, equipped with a screw cap with a hole in the center (Pierce #13075 or equivalent). Detergent wash, rinse with tap and distilled water, and dry at 105 °C before use.
5.1.2 Septum - Teflon-faced silicone (Pierce #12722 or equivalent). Detergent wash, rinse with tap and distilled water, and dry at 105 °C for 1 h before use.
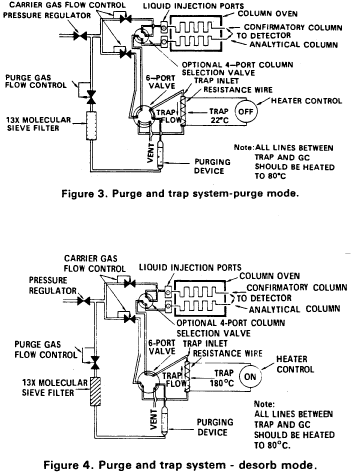
5.2 Purge and trap system - The purge and trap system consists of three separate pieces of equipment: a purging device, trap, and desorber. Several complete systems are now commercially available.
5.2.1 The purging device must be designed to accept 5-mL samples with a water column at least 3 cm deep. The gaseous head space between the water column and the trap must have a total volume of less than 15 mL. The purge gas must pass through the water column as finely divided bubbles with a diameter of less than 3 mm at the origin. The purge gas must be introduced no more than 5 mm from the base of the water column. The purging device illustrated in Figure 1 meets these design criteria.
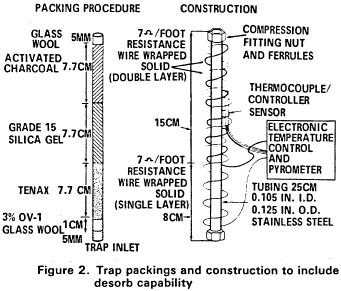
5.2.2 The trap must be at least 25 cm long and have an inside diameter of at least 0.105 in. The trap must be packed to contain the following minimum lengths of adsorbents: 1.0 cm of methyl silicone coated packing (Section 6.3.3), 7.7 cm of 2,6-diphenylene oxide polymer (Section 6.3.2), 7.7 cm of silica gel (Section 6.3.4), 7.7 cm of coconut charcoal (Section 6.3.1). If it is not necessary to analyze for dichlorodifluoromethane, the charcoal can be eliminated, and the polymer section lengthened to 15 cm. The minimum specifications for the trap are illustrated in Figure 2.
5.2.3 The desorber must be capable of rapidly heating the trap to 180 °C. The polymer section of the trap should not be heated higher than 180 °C and the remaining sections should not exceed 200 °C. The desorber illustrated in Figure 2 meets these design criteria.
5.2.4 The purge and trap system may be assembled as a separate unit or be coupled to a gas chromatograph as illustrated in Figures 3 and 4.
5.3 Gas chromatograph - An analytical system complete with a temperature programmable gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.3.1 Column 1 - 8 ft long × 0.1 in. ID stainless steel or glass, packed with 1% SP-1000 on Carbopack B (60/80 mesh) or equivalent. This column was used to develop the method performance statements in Section 12. Guidelines for the use of alternate column packings are provided in Section 10.1.
5.3.2 Column 2 - 6 ft long × 0.1 in. ID stainless steel or glass, packed with chemically bonded n-octane on Porasil-C (100/120 mesh) or equivalent.
5.3.3 Detector - Electrolytic conductivity or microcoulometric detector. These types of detectors have proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1). The electrolytic conductivity detector was used to develop the method performance statements in Section 12. Guidelines for the use of alternate detectors are provided in Section 10.1.
5.4 Syringes - 5-mL glass hypodermic with Luerlok tip (two each), if applicable to the purging device.
5.5 Micro syringes - 25-µL, 0.006 in. ID needle.
5.6 Syringe valve - 2-way, with Luer ends (three each).
5.7 Syringe - 5-mL, gas-tight with shut-off valve.
5.8 Bottle - 15-mL, screw-cap, with Teflon cap liner.
5.9 Balance - Analytical, capable of accurately weighing 0.0001 g.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.1.1 Reagent water can be generated by passing tap water through a carbon filter bed containing about 1 lb of activated carbon (Filtrasorb-300, Calgon Corp., or equivalent).
6.1.2 A water purification system (Millipore Super-Q or equivalent) may be used to generate reagent water.
6.1.3 Reagent water may also be prepared by boiling water for 15 min. Subsequently, while maintaining the temperature at 90 °C, bubble a contaminant-free inert gas through the water for 1 h. While still hot, transfer the water to a narrow mouth screw-cap bottle and seal with a Teflon-lined septum and cap.
6.2 Sodium thiosulfate - (ACS) Granular.
6.3 Trap Materials:
6.3.1 Coconut charcoal - 6/10 mesh sieved to 26 mesh, Barnabey Cheney, CA-580-26 lot # M-2649 or equivalent.
6.3.2 2,6-Diphenylene oxide polymer - Tenax, (60/80 mesh), chromatographic grade or equivalent.
6.3.3 Methyl silicone packing - 3% OV-1 on Chromosorb-W (60/80 mesh) or equivalent.
6.3.4 Silica gel - 35/60 mesh, Davison, grade-15 or equivalent.
6.4 Methanol - Pesticide quality or equivalent.
6.5 Stock standard solutions - Stock standard solutions may be prepared from pure standard materials or purchased as certified solutions. Prepare stock standard solutions in methanol using assayed liquids or gases as appropriate. Because of the toxicity of some of the organohalides, primary dilutions of these materials should be prepared in a hood. A NIOSH/MESA approved toxic gas respirator should be used when the analyst handles high concentrations of such materials.
6.5.1 Place about 9.8 mL of methanol into a 10-mL ground glass stoppered volumetric flask. Allow the flask to stand, unstoppered, for about 10 min or until all alcohol wetted surfaces have dried. Weigh the flask to the learest 0.1 mg.
6.5.2 Add the assayed reference material:
6.5.2.1 Liquid - Using a 100 µL syringe, immediately add two or more drops of assayed reference material to the flask, then reweigh. Be sure that the drops fall directly into the alcohol without contacting the neck of the flask.
6.5.2.2 Gases - To prepare standards for any of the six halocarbons that boil below 30 °C (bromomethane, chloroethane, chloromethane, dichlorodifluoromethane, trichlorofluoromethane, vinyl chloride), fill a 5-mL valved gas-tight syringe with the reference standard to the 5.0-mL mark. Lower the needle to 5 mm above the methanol meniscus. Slowly introduce the reference standard above the surface of the liquid (the heavy gas will rapidly dissolve into the methanol).
6.5.3 Reweigh, dilute to volume, stopper, then mix by inverting the flask several times. Calculate the concentration in µg/µL from the net gain in weight. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the malufacturer or by an independent source.
6.5.4 Transfer the stock standard solution into a Teflon-sealed screw-cap bottle. Store, with minimal headspace, at −10 to −20 °C and protect from light.
6.5.5 Prepare fresh standards weekly for the six gases and 2-chloroethylvinyl ether. All other standards must be replaced after one month, or sooner if comparison with check standards indicates a problem.
6.6 Secondary dilution standards - Using stock standard solutions, prepare secondary dilution standards in methanol that contain the compounds of interest, either singly or mixed together. The secondary dilution standards should be prepared at concentrations such that the aqueous calibration standards prepared in Section 7.3.1 or 7.4.1 will bracket the working range of the analytical system. Secondary dilution standards should be stored with minimal headspace and should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.7 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Assemble a purge and trap system that meets the specifications in Section 5.2. Condition the trap overnight at 180 °C by backflushing with an inert gas flow of at least 20 mL/min. Condition the trap for 10 min once daily prior to use.
7.2 Connect the purge and trap system to a gas chromatograph. The gas chromatograph must be operated using temperature and flow rate conditions equivalent to those given in Table 1. Calibrate the purge and trap-gas chromatographic system using either the external standard technique (Section 7.3) or the internal standard technique (Section 7.4).
7.3 External standard calibration procedure:
7.3.1 Prepare calibration standards at a miminum of three concentration levels for each parameter by carefully adding 20.0 µL of one or more secondary dilution standards to 100, 500, or 1000 µL of reagent water. A 25-µL syringe with a 0.006 in. ID needle should be used for this operation. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector. These aqueous standards can be stored up to 24 h, if held in sealed vials with zero headspace as described in Section 9.2. If not so stored, they must be discarded after 1 h.
7.3.2 Analyze each calibration standard according to Section 10, and tabulate peak height or area responses versus the concentration in the standard. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to concentration (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.4 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples. The compounds recommended for use as surrogate spikes in Section 8.7 have been used successfully as internal standards, because of their generally unique retention times.
7.4.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest as described in Section 7.3.1.
7.4.2 Prepare a spiking solution containing each of the internal standards using the procedures described in Sections 6.5 and 6.6. It is recommended that the secondary dilution standard be prepared at a concentration of 15 µg/mL of each internal standard compound. The addition of 10 µL of this standard to 5.0 mL of sample or calibration standard would be equivalent to 30 µg/L.
7.4.3 Analyze each calibration standard according to Section 10, adding 10 µL of internal standard spiking solution directly to the syringe (Section 10.4). Tabulate peak height or area responses against concentration for each compound and internal standard, and calculate response factors (RF) for each compound using Equation 1.
Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard. Cs = Concentration of the parameter to be measured. If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.7.5 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of a QC check sample.
7.5.1 Prepare the QC check sample as described in Section 8.2.2.
7.5.2 Analyze the QC check sample according to Section 10.
7.5.3 For each parameter, compare the response (Q) with the corresponding calibration acceptance criteria found in Table 2. If the responses for all parameters of interest fall within the designated ranges, analysis of actual samples can begin. If any individual Q falls outside the range, proceed according to Section 7.5.4.
Note:The large number of parameters in Table 2 present a substantial probability that one or more will not meet the calibration acceptance criteria when all parameters are analyzed.
7.5.4 Repeat the test only for those parameters that failed to meet the calibration acceptance criteria. If the response for a parameter does not fall within the range in this second test, a new calibration curve, calibration factor, or RF must be prepared for that parameter according to Section 7.3 or 7.4.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Section 10.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Each day, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system are under control.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest at a concentration of 10 µg/mL in methanol. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Prepare a QC check sample to contain 20 µg/L of each parameter by adding 200 µL of QC check sample concentrate to 100 mL of reagent water.
8.2.3 Analyze four 5-mL aliquots of the well-mixed QC check sample according to Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter of interest using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, then the system performance is unacceptable for that parameter.
Note:The large number of parameters in Table 2 present a substantial probability that one or more will fail at least one of the acceptance criteria when all parameters are analyzed.
8.2.6 When one or more of the parameters tested fail at least one of the acceptance criteria, the analyst must proceed according to Section 8.2.6.1 or 8.2.6.2.
8.2.6.1 Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.3.
8.2.6.2 Beginning with Section 8.2.3, repeat the test only for those parameters that failed to meet criteria. Repeated failure, however, will confirm a general problem with the measurement system. If this occurs, locate and correct the source of the problem and repeat the test for all compounds of interest beginning with Section 8.2.3.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at 20 µg/L or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.2 Analyze one 5-mL sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second 5-mL sample aliquot with 10 µL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100(A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 7 If spiking was performed at a concentration lower than 20 µg/L, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T)±2.44(100 S′/T)%. 7
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory. If the entire list of parameters in Table 2 must be measured in the sample in Section 8.3, the probability that the analysis of a QC check standard will be required is high. In this case the QC check standard should be routinely analyzed with the spiked sample.
8.4.1 Prepare the QC check standard by adding 10 µL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 5 mL of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 2. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If p = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
8.7 The analyst should monitor both the performance of the analytical system and the effectiveness of the method in dealing with each sample matrix by spiking each sample, standard, and reagent water blank with surrogate halocarbons. A combination of bromochloromethane, 2-bromo-1-chloropropane, and 1,4-dichlorobutane is recommended to encompass the range of the temperature program used in this method. From stock standard solutions prepared as in Section 6.5, add a volume to give 750 µg of each surrogate to 45 mL of reagent water contained in a 50-mL volumetric flask, mix and dilute to volume for a concentration of 15 ng/µL. Add 10 µL of this surrogate spiking solution directly into the 5-mL syringe with every sample and reference standard analyzed. Prepare a fresh surrogate spiking solution on a weekly basis. If the internal standard calibration procedure is being used, the surrogate compounds may be added directly to the internal standard spiking solution (Section 7.4.2).
9. Sample Collection, Preservation, and Handling9.1 All samples must be iced or refrigerated from the time of collection until analysis. If the sample contains free or combined chlorine, add sodium thiosulfate preservative (10 mg/40 mL is sufficient for up to 5 ppm Cl2) to the empty sample bottle just prior to shipping to the sampling site. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine. 8 Field test kits are available for this purpose.
9.2 Grab samples must be collected in glass containers having a total volume of at least 25 mL. Fill the sample bottle just to overflowing in such a manner that no air bubbles pass through the sample as the bottle is being filled. Seal the bottle so that no air bubbles are entrapped in it. If preservative has been added, shake vigorously for 1 min. Maintain the hermetic seal on the sample bottle until time of analysis.
9.3 All samples must be analyzed within 14 days of collection. 3
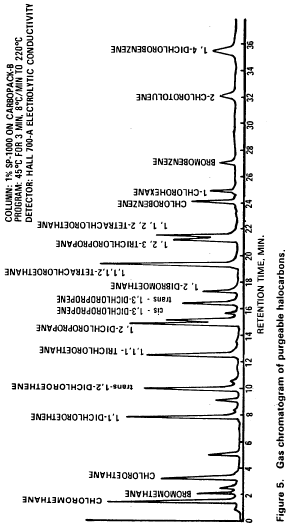
10. Procedure10.1 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are estimated retention times and MDL that can be achieved under these conditions. An example of the separations achieved by Column 1 is shown in Figure 5. Other packed columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
10.2 Calibrate the system daily as described in Section 7.
10.3 Adjust the purge gas (nitrogen or helium) flow rate to 40 mL/min. Attach the trap inlet to the purging device, and set the purge and trap system to purge (Figure 3). Open the syringe valve located on the purging device sample introduction needle.
10.4 Allow the sample to come to ambient temperature prior to introducing it to the syringe. Remove the plunger from a 5-mL syringe and attach a closed syringe valve. Open the sample bottle (or standard) and carefully pour the sample into the syringe barrel to just short of overflowing. Replace the syringe plunger and compress the sample. Open the syringe valve and vent any residual air while adjusting the sample volume to 5.0 mL. Since this process of taking an aliquot destroys the validity of the sample for future analysis, the analyst should fill a second syringe at this time to protect against possible loss of data. Add 10.0 µL of the surrogate spiking solution (Section 8.7) and 10.0 µL of the internal standard spiking solution (Section 7.4.2), if applicable, through the valve bore, then close the valve.
10.5 Attach the syringe-syringe valve assembly to the syringe valve on the purging device. Open the syringe valves and inject the sample into the purging chamber.
10.6 Close both valves and purge the sample for 11.0 ±0.1 min at ambient temperature.
10.7 After the 11-min purge time, attach the trap to the chromatograph, adjust the purge and trap system to the desorb mode (Figure 4), and begin to temperature program the gas chromatograph. Introduce the trapped materials to the GC column by rapidly heating the trap to 180 °C while backflushing the trap with an inert gas between 20 and 60 mL/min for 4 min. If rapid heating of the trap cannot be achieved, the GC column must be used as a secondary trap by cooling it to 30 °C (subambient temperature, if poor peak geometry or random retention time problems persist) instead of the initial program temperature of 45 °C
10.8 While the trap is being desorbed into the gas chromatograph, empty the purging chamber using the sample introduction syringe. Wash the chamber with two 5-mL flushes of reagent water.
10.9 After desorbing the sample for 4 min, recondition the trap by returning the purge and trap system to the purge mode. Wait 15 s then close the syringe valve on the purging device to begin gas flow through the trap. The trap temperature should be maintained at 180 °C After approximately 7 min, turn off the trap heater and open the syringe valve to stop the gas flow through the trap. When the trap is cool, the next sample can be analyzed.
10.10 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
10.11 If the response for a peak exceeds the working range of the system, prepare a dilution of the sample with reagent water from the aliquot in the second syringe and reanalyze.
11. Calculations11.1 Determine the concentration of individual compounds in the sample.
11.1.1 If the external standard calibration procedure is used, calculate the concentration of the parameter being measured from the peak response using the calibration curve or calibration factor determined in Section 7.3.2.
11.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.4.3 and Equation 2.
Equation 2 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard.11.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
12. Method Performance12.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentration listed in Table 1 were obtained using reagent water. 11. Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
12.2 This method is recommended for use in the concentration range from the MDL to 1000 × MDL. Direct aqueous injection techniques should be used to measure concentration levels above 1000 × MDL.
12.3 This method was tested by 20 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 8.0 to 500 µg/L. 9 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. 40 CFR part 136, appendix B.
2. Bellar, T.A., and Lichtenberg, J.J. “Determining Volatile Organics at Microgram-per-Litre-Levels by Gas Chromatography,” Journal of the American Water Works Association, 66, 739 (1974).
3. Bellar, T.A., and Lichtenberg, J.J. “Semi-Automated Headspace Analysis of Drinking Waters and Industrial Waters for Purgeable Volatile Organic Compounds,” Proceedings from Symposium on Measurement of Organic Pollutants in Water and Wastewater, American Society for Testing and Materials, STP 686, C.E. Van Hall, editor, 1978.
4. “Carcinogens - Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
5. “OSHA Safety and Health Standards, General Industry” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
7. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
8. “Methods 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric, DPD) for Chlorine, Total Residual,” Methods for Chemical Analysis of Water and Wastes, EPA 600/4-79-020, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1979.
9. “EPA Method Study 24, Method 601 - Purgeable Halocarbons by the Purge and Trap Method,” EPA 600/4-84-064, National Technical Information Service, PB84-212448, Springfield, Virginia 22161, July 1984.
10. “Method Validation Data for EPA Method 601,” Memorandum from B. Potter, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, November 10, 1983.
11. Bellar, T. A., Unpublished data, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, 1981.
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Method detection limit (µg/L) | |
|---|---|---|---|
| Column 1 | Column 2 | ||
| Chloromethane | 1.50 | 5.28 | 0.08 |
| Bromomethane | 2.17 | 7.05 | 1.18 |
| Dichlorodifluoromethane | 2.62 | nd | 1.81 |
| Vinyl chloride | 2.67 | 5.28 | 0.18 |
| Chloroethane | 3.33 | 8.68 | 0.52 |
| Methylene chloride | 5.25 | 10.1 | 0.25 |
| Trichlorofluoromethane | 7.18 | nd | nd |
| 1,1-Dichloroethene | 7.93 | 7.72 | 0.13 |
| 1,1-Dichloroethane | 9.30 | 12.6 | 0.07 |
| trans-1,2-Dichloroethene | 10.1 | 9.38 | 0.10 |
| Chloroform | 10.7 | 12.1 | 0.05 |
| 1,2-Dichloroethane | 11.4 | 15.4 | 0.03 |
| 1,1,1-Trichloroethane | 12.6 | 13.1 | 0.03 |
| Carbon tetrachloride | 13.0 | 14.4 | 0.12 |
| Bromodichloromethane | 13.7 | 14.6 | 0.10 |
| 1,2-Dichloropropane | 14.9 | 16.6 | 0.04 |
| cis-1,3-Dichloropropene | 15.2 | 16.6 | 0.34 |
| Trichloroethene | 15.8 | 13.1 | 0.12 |
| Dibromochloromethane | 16.5 | 16.6 | 0.09 |
| 1,1,2-Trichloroethane | 16.5 | 18.1 | 0.02 |
| trans-1,3-Dichloropropene | 16.5 | 18.0 | 0.20 |
| 2-Chloroethylvinyl ether | 18.0 | nd | 0.13 |
| Bromoform | 19.2 | 19.2 | 0.20 |
| 1,1,2,2-Tetrachloroethane | 21.6 | nd | 0.03 |
| Tetrachloroethene | 21.7 | 15.0 | 0.03 |
| Chlorobenzene | 24.2 | 18.8 | 0.25 |
| 1,3-Dichlorobenzene | 34.0 | 22.4 | 0.32 |
| 1,2-Dichlorobenzene | 34.9 | 23.5 | 0.15 |
| 1,4-Dichlorobenzene | 35.4 | 22.3 | 0.24 |
Column 1 conditions: Carbopack B (60/80 mesh) coated with 1% SP-1000 packed in an 8 ft × 0.1 in. ID stainless steel or glass column with helium carrier gas at 40 mL/min flow rate. Column temperature held at 45 °C for 3 min then programmed at 8 °C/min to 220 °C and held for 15 min.
Column 2 conditions: Porisil-C (100/120 mesh) coated with n-octane packed in a 6 ft × 0.1 in. ID stainless steel or glass column with helium carrier gas at 40 mL/min flow rate. Column temperature held at 50 °C for 3 min then programmed at 6 °C/min to 170 °C and held for 4 min.
nd = not determined.
Table 2 - Calibration and QC Acceptance Criteria - Method 601 a
| Parameter | Range for Q (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range P, Ps (%) |
|---|---|---|---|---|
| Bromodichloromethane | 15.2-24.8 | 4.3 | 10.7-32.0 | 42-172 |
| Bromoform | 14.7-25.3 | 4.7 | 5.0-29.3 | 13-159 |
| Bromomethane | 11.7-28.3 | 7.6 | 3.4-24.5 | D-144 |
| Carbon tetrachloride | 13.7-26.3 | 5.6 | 11.8-25.3 | 43-143 |
| Chlorobenzene | 14.4-25.6 | 5.0 | 10.2-27.4 | 38-150 |
| Chloroethane | 15.4-24.6 | 4.4 | 11.3-25.2 | 46-137 |
| 2-Chloroethylvinyl ether | 12.0-28.0 | 8.3 | 4.5-35.5 | 14-186 |
| Chloroform | 15.0-25.0 | 4.5 | 12.4-24.0 | 49-133 |
| Chloromethane | 11.9-28.1 | 7.4 | D-34.9 | D-193 |
| Dibromochloromethane | 13.1-26.9 | 6.3 | 7.9-35.1 | 24-191 |
| 1,2-Dichlorobenzene | 14.0-26.0 | 5.5 | 1.7-38.9 | D-208 |
| 1,3-Dichlorobenzene | 9.9-30.1 | 9.1 | 6.2-32.6 | 7-187 |
| 1,4-Dichlorobenzene | 13.9-26.1 | 5.5 | 11.5-25.5 | 42-143 |
| 1,1-Dichloroethane | 16.8-23.2 | 3.2 | 11.2-24.6 | 47-132 |
| 1,2-Dichloroethane | 14.3-25.7 | 5.2 | 13.0-26.5 | 51-147 |
| 1,1-Dichloroethene | 12.6-27.4 | 6.6 | 10.2-27.3 | 28-167 |
| trans-1,2-Dichloroethene | 12.8-27.2 | 6.4 | 11.4-27.1 | 38-155 |
| 1,2-Dichloropropane | 14.8-25.2 | 5.2 | 10.1-29.9 | 44-156 |
| cis-1,3-Dichloropropene | 12.8-27.2 | 7.3 | 6.2-33.8 | 22-178 |
| trans-1,3-Dichloropropene | 12.8-27.2 | 7.3 | 6.2-33.8 | 22-178 |
| Methylene chloride | 15.5-24.5 | 4.0 | 7.0-27.6 | 25-162 |
| 1,1,2,2-Tetrachloroethane | 9.8-30.2 | 9.2 | 6.6-31.8 | 8-184 |
| Tetrachloroethene | 14.0-26.0 | 5.4 | 8.1-29.6 | 26-162 |
| 1,1,1-Trichloroethane | 14.2-25.8 | 4.9 | 10.8-24.8 | 41-138 |
| 1,1,2-Trichloroethane | 15.7-24.3 | 3.9 | 9.6-25.4 | 39-136 |
| Trichloroethene | 15.4-24.6 | 4.2 | 9.2-26.6 | 35-146 |
| Trichlorofluoromethane | 13.3-26.7 | 6.0 | 7.4-28.1 | 21-156 |
| Vinyl chloride | 13.7-26.3 | 5.7 | 8.2-29.9 | 28-163 |
a Criteria were calculated assuming a QC check sample concentration of 20 µg/L.
Q = Concentration measured in QC check sample, in µg/L (Section 7.5.3).
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
D = Detected; result must be greater than zero.
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 601
| Parameter | Accuracy, as recovery, X′ (µg/L) | Single analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| Bromodichloromethane | 1.12C−1.02 | 0.11X + 0.04 | 0.20X + 1.00 |
| Bromoform | 0.96C−2.05 | 0.12X + 0.58 | 0.21X + 2.41 |
| Bromomethane | 0.76C−1.27 | 0.28X + 0.27 | 0.36X + 0.94 |
| Carbon tetrachloride | 0.98C−1.04 | 0.15X + 0.38 | 0.20X + 0.39 |
| Chlorobenzene | 1.00C−1.23 | 0.15X −0.02 | 0.18X + 1.21 |
| Choroethane | 0.99C−1.53 | 0.14X −0.13 | 0.17X + 0.63 |
| 2-Chloroethylvinyl ether a | 1.00C | 0.20X | 0.35X |
| Chloroform | 0.93C−0.39 | 0.13X + 0.15 | 0.19X −0.02 |
| Chloromethane | 0.77C + 0.18 | 0.28X −0.31 | 0.52X + 1.31 |
| Dibromochloromethane | 0.94C + 2.72 | 0.11X + 1.10 | 0.24X + 1.68 |
| 1,2-Dichlorobenzene | 0.93C + 1.70 | 0.20X + 0.97 | 0.13X + 6.13 |
| 1,3-Dichlorobenzene | 0.95C + 0.43 | 0.14X + 2.33 | 0.26X + 2.34 |
| 1,4-Dichlorobenzene | 0.93C−0.09 | 0.15X + 0.29 | 0.20X + 0.41 |
| 1,1-Dichloroethane | 0.95C−1.08 | 0.09X + 0.17 | 0.14X + 0.94 |
| 1,2-Dichloroethane | 1.04C−1.06 | 0.11X + 0.70 | 0.15X + 0.94 |
| 1,1-Dichloroethene | 0.98C−0.87 | 0.21X −0.23 | 0.29X −0.40 |
| trans-1,2-Dichloroethene | 0.97C−0.16 | 0.11X + 1.46 | 0.17X + 1.46 |
| 1,2-Dichloropropane a | 1.00C | 0.13X | 0.23X |
| cis-1,3-Dichloropropene a | 1.00C | 0.18X | 0.32X |
| trans-1,3-Dichloropropene a | 1.00C | 0.18X | 0.32X |
| Methylene chloride | 0.91C−0.93 | 0.11X + 0.33 | 0.21X + 1.43 |
| 1,1,2,2-Tetrachloroethene | 0.95C + 0.19 | 0.14X + 2.41 | 0.23X + 2.79 |
| Tetrachloroethene | 0.94C + 0.06 | 0.14X + 0.38 | 0.18X + 2.21 |
| 1,1,1-Trichloroethane | 0.90C−0.16 | 0.15X + 0.04 | 0.20X + 0.37 |
| 1,1,2-Trichloroethane | 0.86C + 0.30 | 0.13X −0.14 | 0.19X + 0.67 |
| Trichloroethene | 0.87C + 0.48 | 0.13X −0.03 | 0.23X + 0.30 |
| Trichlorofluoromethane | 0.89C−0.07 | 0.15X + 0.67 | 0.26X + 0.91 |
| Vinyl chloride | 0.97C−0.36 | 0.13X + 0.65 | 0.27X + 0.40 |
X ′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sn′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S 1 = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
a Estimates based upon the performance in a single laboratory. 10

 Method 602 -
Purgeable Aromatics 1. Scope and Application
Method 602 -
Purgeable Aromatics 1. Scope and Application
1.1 This method covers the determination of various purgeable aromatics. The following parameters may be determined by this method:
| Parameter | STORET No. | CAS No. |
|---|---|---|
| Benzene | 34030 | 71-43-2 |
| Chlorobenzene | 34301 | 108-90-7 |
| 1,2-Dichlorobenzene | 34536 | 95-50-1 |
| 1,3-Dichlorobenzene | 34566 | 541-73-1 |
| 1,4-Dichlorobenzene | 34571 | 106-46-7 |
| Ethylbenzene | 34371 | 100-41-4 |
| Toluene | 34010 | 108-88-3 |
1.2 This is a purge and trap gas chromatographic (GC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compounds above, compound identifications should be supported by at least one additional qualitative technique. This method describes analytical conditions for a second gas chromatographic column that can be used to confirm measurements made with the primary column. Method 624 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for all of the parameters listed above.
1.3 The method detection limit (MDL, defined in Section 12.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.4 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.5 This method is restricted to use by or under the supervision of analysts experienced in the operation of a purge and trap system and a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 An inert gas is bubbled through a 5-mL water sample contained in a specially-designed purging chamber at ambient temperature. The aromatics are efficiently transferred from the aqueous phase to the vapor phase. The vapor is swept through a sorbent trap where the aromatics are trapped. After purging is completed, the trap is heated and backflushed with the inert gas to desorb the aromatics onto a gas chromatographic column. The gas chromatograph is temperature programmed to separate the aromatics which are then detected with a photoionization detector. 2 3
2.2 The method provides an optional gas chromatographic column that may be helpful in resolving the compounds of interest from interferences that may occur.
3. Interferences3.1 Impurities in the purge gas and organic compounds outgassing from the plumbing ahead of the trap account for the majority of contamination problems. The analytical system must be demonstrated to be free from contamination under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3. The use of non-Teflon plastic tubing, non-Teflon thread sealants, or flow controllers with rubber components in the purge and trap system should be avoided.
3.2 Samples can be contaminated by diffusion of volatile organics through the septum seal into the sample during shipment and storage. A field reagent blank prepared from reagent water and carried through the sampling and handling protocol can serve as a check on such contamination.
3.3 Contamination by carry-over can occur whenever high level and low level samples are sequentially analyzed. To reduce carry-over, the purging device and sample syringe must be rinsed with reagent water between sample analyses. Whenever an unusually concentrated sample is encountered, it should be followed by an analysis of reagent water to check for cross contamination. For samples containing large amounts of water-soluble materials, suspended solids, high boiling compounds or high aromatic levels, it may be necessary to wash the purging device with a detergent solution, rinse it with distilled water, and then dry it in an oven at 105 °C between analyses. The trap and other parts of the system are also subject to contamination; therefore, frequent bakeout and purging of the entire system may be required.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 4 6 for the information of the analyst.
4.2 The following parameters covered by this method have been tentatively classified as known or suspected, human or mammalian carcinogens: benzene and 1,4-dichlorobenzene. Primary standards of these toxic compounds should be prepared in a hood. A NIOSH/MESA approved toxic gas respirator should be worn when the analyst handles high concentrations of these toxic compounds.
5. Apparatus and Materials5.1 Sampling equipment, for discrete sampling.
5.1.1 Vial]25-mL capacity or larger, equipped with a screw cap with a hole in the center (Pierce #13075 or equivalent). Detergent wash, rinse with tap and distilled water, and dry at 105 °C before use.
5.1.2 Septum - Teflon-faced silicone (Pierce #12722 or equivalent). Detergent wash, rinse with tap and distilled water, and dry at 105 °C for 1 h before use.
5.2 Purge and trap system - The purge and trap system consists of three separate pieces of equipment: A purging device, trap, and desorber. Several complete systems are now commercially available.
5.2.1 The purging device must be designed to accept 5-mL samples with a water column at least 3 cm deep. The gaseous head space between the water column and the trap must have a total volume of less than 15 mL. The purge gas must pass through the water column as finely divided bubbles with a diameter of less than 3 mm at the origin. The purge gas must be introduced no more than 5 mm from the base of the water column. The purging device illustrated in Figure 1 meets these design criteria.
5.2.2 The trap must be at least 25 cm long and have an inside diameter of at least 0.105 in.
5.2.2.1 The trap is packed with 1 cm of methyl silicone coated packing (Section 6.4.2) and 23 cm of 2,6-diphenylene oxide polymer (Section 6.4.1) as shown in Figure 2. This trap was used to develop the method performance statements in Section 12.
5.2.2.2 Alternatively, either of the two traps described in Method 601 may be used, although water vapor will preclude the measurement of low concentrations of benzene.
5.2.3 The desorber must be capable of rapidly heating the trap to 180 °C. The polymer section of the trap should not be heated higher than 180 °C and the remaining sections should not exceed 200 °C. The desorber illustrated in Figure 2 meets these design criteria.
5.2.4 The purge and trap system may be assembled as a separate unit or be coupled to a gas chromatograph as illustrated in Figures 3, 4, and 5.
5.3 Gas chromatograph - An analytical system complete with a temperature programmable gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.3.1 Column 1 - 6 ft long × 0.082 in. ID stainless steel or glass, packed with 5% SP-1200 and 1.75% Bentone-34 on Supelcoport (100/120 mesh) or equivalent. This column was used to develop the method performance statements in Section 12. Guidelines for the use of alternate column packings are provided in Section 10.1.
5.3.2 Column 2 - 8 ft long × 0.1 in ID stainless steel or glass, packed with 5% 1,2,3-Tris(2-cyanoethoxy)propane on Chromosorb W-AW (60/80 mesh) or equivalent.
5.3.3 Detector - Photoionization detector (h-Nu Systems, Inc. Model PI-51-02 or equivalent). This type of detector has been proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1), and was used to develop the method performance statements in Section 12. Guidelines for the use of alternate detectors are provided in Section 10.1.
5.4 Syringes - 5-mL glass hypodermic with Luerlok tip (two each), if applicable to the purging device.
5.5 Micro syringes - 25-µL, 0.006 in. ID needle.
5.6 Syringe valve - 2-way, with Luer ends (three each).
5.7 Bottle - 15-mL, screw-cap, with Teflon cap liner.
5.8 Balance - Analytical, capable of accurately weighing 0.0001 g.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.1.1 Reagent water can be generated by passing tap water through a carbon filter bed containing about 1 lb of activated carbon (Filtrasorb-300, Calgon Corp., or equivalent).
6.1.2 A water purification system (Millipore Super-Q or equivalent) may be used to generate reagent water.
6.1.3 Reagent water may also be prepared by boiling water for 15 min. Subsequently, while maintaining the temperature at 90 °C, bubble a contaminant-free inert gas through the water for 1 h. While still hot, transfer the water to a narrow mouth screw-cap bottle and seal with a Teflon-lined septum and cap.
6.2 Sodium thiosulfate - (ACS) Granular.
6.3 Hydrochloric acid (1 + 1) - Add 50 mL of concentrated HCl (ACS) to 50 mL of reagent water.
6.4 Trap Materials:
6.4.1 2,6-Diphenylene oxide polymer - Tenax, (60/80 mesh), chromatographic grade or equivalent.
6.4.2 Methyl silicone packing - 3% OV-1 on Chromosorb-W (60/80 mesh) or equivalent.
6.5 Methanol - Pesticide quality or equivalent.
6.6 Stock standard solutions - Stock standard solutions may be prepared from pure standard materials or purchased as certified solutions. Prepare stock standard solutions in methanol using assayed liquids. Because of the toxicity of benzene and 1,4-dichlorobenzene, primary dilutions of these materials should be prepared in a hood. A NIOSH/MESA approved toxic gas respirator should be used when the analyst handles high concentrations of such materials.
6.6.1 Place about 9.8 mL of methanol into a 10-mL ground glass stoppered volumetric flask. Allow the flask to stand, unstoppered, for about 10 min or until all alcohol wetted surfaces have dried. Weigh the flask to the nearest 0.1 mg.
6.6.2 Using a 100-µL syringe, immediately add two or more drops of assayed reference material to the flask, then reweigh. Be sure that the drops fall directly into the alcohol without contacting the neck of the flask.
6.6.3 Reweigh, dilute to volume, stopper, then mix by inverting the flask several times. Calculate the concentration in µg/µL from the net gain in weight. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.6.4 Transfer the stock standard solution into a Teflon-sealed screw-cap bottle. Store at 4 °C and protect from light.
6.6.5 All standards must be replaced after one month, or sooner if comparison with check standards indicates a problem.
6.7 Secondary dilution standards - Using stock standard solutions, prepare secondary dilution standards in methanol that contain the compounds of interest, either singly or mixed together. The secondary dilution standards should be prepared at concentrations such that the aqueous calibration standards prepared in Section 7.3.1 or 7.4.1 will bracket the working range of the analytical system. Secondary solution standards must be stored with zero headspace and should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.8 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Assemble a purge and trap system that meets the specifications in Section 5.2. Condition the trap overnight at 180 °C by backflushing with an inert gas flow of at least 20 mL/min. Condition the trap for 10 min once daily prior to use.
7.2 Connect the purge and trap system to a gas chromatograph. The gas chromatograph must be operated using temperature and flow rate conditions equivalent to those given in Table 1. Calibrate the purge and trap-gas chromatographic system using either the external standard technique (Section 7.3) or the internal standard technique (Section 7.4).
7.3 External standard calibration procedure:
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter by carefully adding 20.0 µL of one or more secondary dilution standards to 100, 500, or 1000 mL of reagent water. A 25-µL syringe with a 0.006 in. ID needle should be used for this operation. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector. These aqueous standards must be prepared fresh daily.
7.3.2 Analyze each calibration standard according to Section 10, and tabulate peak height or area responses versus the concentration in the standard. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to concentration (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.4 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples. The compound, α,α,α,-trifluorotoluene, recommended as a surrogate spiking compound in Section 8.7 has been used successfully as an internal standard.
7.4.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest as described in Section 7.3.1.
7.4.2 Prepare a spiking solution containing each of the internal standards using the procedures described in Sections 6.6 and 6.7. It is recommended that the secondary dilution standard be prepared at a concentration of 15 µg/mL of each internal standard compound. The addition of 10 µl of this standard to 5.0 mL of sample or calibration standard would be equivalent to 30 µg/L.
7.4.3 Analyze each calibration standard according to Section 10, adding 10 µL of internal standard spiking solution directly to the syringe (Section 10.4). Tabulate peak height or area responses against concentration for each compound and internal standard, and calculate response factors (RF) for each compound using Equation 1.
RF = (As)(Cis (Ais)(Cs) Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard Cs = Concentration of the parameter to be measured. If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.7.5 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of a QC check sample.
7.5.1 Prepare the QC check sample as described in Section 8.2.2.
7.5.2 Analyze the QC check sample according to Section 10.
7.5.3 For each parameter, compare the response (Q) with the corresponding calibration acceptance criteria found in Table 2. If the responses for all parameters of interest fall within the designated ranges, analysis of actual samples can begin. If any individual Q falls outside the range, a new calibration curve, calibration factor, or RF must be prepared for that parameter according to Section 7.3 or 7.4.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Section 10.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Each day, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system are under control.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest at a concentration of 10 µg/mL in methanol. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Prepare a QC check sample to contain 20 µg/L of each parameter by adding 200 µL of QC check sample concentrate to 100 mL of reagant water.
8.2.3 Analyze four 5-mL aliquots of the well-mixed QC check sample according to Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter of interest using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter.
Note:The large number of parameters in Table 2 present a substantial probability that one or more will fail at least one of the acceptance criteria when all parameters are analyzed.
8.2.6 When one or more of the parameters tested fail at least one of the acceptance criteria, the analyst must proceed according to Section 8.2.6.1 or 8.2.6.2.
8.2.6.1 Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.3.
8.2.6.2 Beginning with Section 8.2.3, repeat the test only for those parameters that failed to meet criteria. Repeated failure, however, will confirm a general problem with the measurement system. If this occurs, locate and correct the source of the problem and repeat the test for all compounds of interest beginning with Section 8.2.3.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at 20 µg/L or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.2 Analyze one 5-mL sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second 5-mL sample aliquot with 10 µL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100(A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 7 If spiking was performed at a concentration lower than 20 µg/L, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T) ±2.44(100 S′/T)%. 7
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 10 µL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 5 mL of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 2. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
8.7 The analyst should monitor both the performance of the analytical system and the effectiveness of the method in dealing with each sample matrix by spiking each sample, standard, and reagent water blank with surrogate compounds (e.g. α, α, α,-trifluorotoluene) that encompass the range of the temperature program used in this method. From stock standard solutions prepared as in Section 6.6, add a volume to give 750 µg of each surrogate to 45 mL of reagent water contained in a 50-mL volumetric flask, mix and dilute to volume for a concentration of 15 mg/µL. Add 10 µL of this surrogate spiking solution directly into the 5-mL syringe with every sample and reference standard analyzed. Prepare a fresh surrogate spiking solution on a weekly basis. If the internal standard calibration procedure is being used, the surrogate compounds may be added directly to the internal standard spiking solution (Section 7.4.2).
9. Sample Collection, Preservation, and Handling9.1 The samples must be iced or refrigerated from the time of collection until analysis. If the sample contains free or combined chlorine, add sodium thiosulfate preservative (10 mg/40 mL is sufficient for up to 5 ppm Cl2) to the empty sample bottle just prior to shipping to the sampling site. EPA Method 330.4 or 330.5 may be used for measurement of residual chlorine. 8 Field test kits are available for this purpose.
9.2 Collect about 500 mL of sample in a clean container. Adjust the pH of the sample to about 2 by adding 1 + 1 HCl while stirring. Fill the sample bottle in such a manner that no air bubbles pass through the sample as the bottle is being filled. Seal the bottle so that no air bubbles are entrapped in it. Maintain the hermetic seal on the sample bottle until time of analysis.
9.3 All samples must be analyzed within 14 days of collection. 3
10. Procedure10.1 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are estimated retention times and MDL that can be achieved under these conditions. An example of the separations achieved by Column 1 is shown in Figure 6. Other packed columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
10.2 Calibrate the system daily as described in Section 7.
10.3 Adjust the purge gas (nitrogen or helium) flow rate to 40 mL/min. Attach the trap inlet to the purging device, and set the purge and trap system to purge (Figure 3). Open the syringe valve located on the purging device sample introduction needle.
10.4 Allow the sample to come to ambient temperature prior to introducing it to the syringe. Remove the plunger from a 5-mL syringe and attach a closed syringe valve. Open the sample bottle (or standard) and carefully pour the sample into the syringe barrel to just short of overflowing. Replace the syringe plunger and compress the sample. Open the syringe valve and vent any residual air while adjusting the sample volume to 5.0 mL. Since this process of taking an aliquot destroys the validity of the sample for future analysis, the analyst should fill a second syringe at this time to protect against possible loss of data. Add 10.0 µL of the surrogate spiking solution (Section 8.7) and 10.0 µL of the internal standard spiking solution (Section 7.4.2), if applicable, through the valve bore, then close the valve.
10.5 Attach the syringe-syringe valve assembly to the syringe valve on the purging device. Open the syringe valves and inject the sample into the purging chamber.
10.6 Close both valves and purge the sample for 12.0 ±0.1 min at ambient temperature.
10.7 After the 12-min purge time, disconnect the purging device from the trap. Dry the trap by maintaining a flow of 40 mL/min of dry purge gas through it for 6 min (Figure 4). If the purging device has no provision for bypassing the purger for this step, a dry purger should be inserted into the device to minimize moisture in the gas. Attach the trap to the chromatograph, adjust the purge and trap system to the desorb mode (Figure 5), and begin to temperature program the gas chromatograph. Introduce the trapped materials to the GC column by rapidly heating the trap to 180 °C while backflushing the trap with an inert gas between 20 and 60 mL/min for 4 min. If rapid heating of the trap cannot be achieved, the GC column must be used as a secondary trap by cooling it to 30 °C (subambient temperature, if poor peak geometry and random retention time problems persist) instead of the initial program temperature of 50 °C.
10.8 While the trap is being desorbed into the gas chromatograph column, empty the purging chamber using the sample introduction syringe. Wash the chamber with two 5-mL flushes of reagent water.
10.9 After desorbing the sample for 4 min, recondition the trap by returning the purge and trap system to the purge mode. Wait 15 s, then close the syringe valve on the purging device to begin gas flow through the trap. The trap temperature should be maintained at 180 °C. After approximately 7 min, turn off the trap heater and open the syringe valve to stop the gas flow through the trap. When the trap is cool, the next sample can be analyzed.
10.10 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
10.11 If the response for a peak exceeds the working range of the system, prepare a dilution of the sample with reagent water from the aliquot in the second syringe and reanalyze.
11. Calculations11.1 Determine the concentration of individual compounds in the sample.
11.1.1 If the external standard calibration procedure is used, calculate the concentration of the parameter being measured from the peak response using the calibration curve or calibration factor determined in Section 7.3.2.
11.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.4.3 and Equation 2.
Equation 2
where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard.11.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
12. Method Performance12.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Table 1 were obtained using reagent water. 9 Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
12.2 This method has been demonstrated to be applicable for the concentration range from the MDL to 100 × MDL. 9 Direct aqueous injection techniques should be used to measure concentration levels above 1000 × MDL.
12.3 This method was tested by 20 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 2.1 to 550 µg/L. 9 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. 40 CFR part 136, appendix B.
2. Lichtenberg, J.J. “Determining Volatile Organics at Microgram-per-Litre-Levels by Gas Chromatography,” Journal American Water Works Association, 66, 739 (1974).
3. Bellar, T.A., and Lichtenberg, J.J. “Semi-Automated Headspace Analysis of Drinking Waters and Industrial Waters for Purgeable Volatile Organic Compounds,” Proceedings of Symposium on Measurement of Organic Pollutants in Water and Wastewater. American Society for Testing and Materials, STP 686, C.E. Van Hall, editor, 1978.
4. “Carcinogens - Working with Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health. Publication No. 77-206, August 1977.
5. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Safety, 3rd Edition, 1979.
7. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3. is two times the value 1.22 derived in this report.)
8.“Methods 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric, DPD) for Chlorine, Total Residual,” Methods for Chemical Analysis of Water and Wastes, EPA-600/4-79-020, U.S. Environmental Protection Agency, Office of Research and Development, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268. March 1979.
9. “EPA Method Study 25, Method 602, Purgeable Aromatics,” EPA 600/4-84-042, National Technical Information Service, PB84-196682, Springfield, Virginia 22161, May 1984.
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Method detection limit (µg/L) | |
|---|---|---|---|
| Column 1 | Column 2 | ||
| Benzene | 3.33 | 2.75 | 0.2 |
| Toluene | 5.75 | 4.25 | 0.2 |
| Ethylbenzene | 8.25 | 6.25 | 0.2 |
| Chlorobenzene | 9.17 | 8.02 | 0.2 |
| 1,4-Dichlorobenzene | 16.8 | 16.2 | 0.3 |
| 1,3-Dichlorobenzene | 18.2 | 15.0 | 0.4 |
| 1,2-Dichlorobenzene | 25.9 | 19.4 | 0.4 |
Column 1 conditions: Supelcoport (100/120 mesh) coated with 5% SP-1200/1.75% Bentone-34 packed in a 6 ft × 0.085 in. ID stainless steel column with helium carrier gas at 36 mL/min flow rate. Column temperature held at 50 °C for 2 min then programmed at 6 °C/min to 90 °C for a final hold.
Column 2 conditions: Chromosorb W-AW (60/80 mesh) coated with 5% 1,2,3-Tris(2-cyanoethyoxy)propane packed in a 6 ft × 0.085 in. ID stainless steel column with helium carrier gas at 30 mL/min flow rate. Column temperature held at 40 °C for 2 min then programmed at 2 °C/min to 100 °C for a final hold.
Table 2 - Calibration and QC Acceptance Criteria - Method 602 a
| Parameter | Range for Q (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps(%) |
|---|---|---|---|---|
| Benzene | 15.4-24.6 | 4.1 | 10.0-27.9 | 39-150 |
| Chlorobenzene | 16.1-23.9 | 3.5 | 12.7-25.4 | 55-135 |
| 1,2-Dichlorobenzene | 13.6-26.4 | 5.8 | 10.6-27.6 | 37-154 |
| 1,3-Dichlorobenzene | 14.5-25.5 | 5.0 | 12.8-25.5 | 50-141 |
| 1,4-Dichlorobenzene | 13.9-26.1 | 5.5 | 11.6-25.5 | 42-143 |
| Ethylbenzene | 12.6-27.4 | 6.7 | 10.0-28.2 | 32-160 |
| Toluene | 15.5-24.5 | 4.0 | 11.2-27.7 | 46-148 |
Q = Concentration measured in QC check sample, in µg/L (Section 7.5.3).
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
Ps, P = Percent recovery measured (Section 8.3.2, Section 8.4.2).
a Criteria were calculated assuming a QC check sample concentration of 20 µg/L.
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 602
| Parameter | Accuracy, as recovery, X′ (µg/L) | Single analyst precision, s′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| Benzene | 0.92C + 0.57 | 0.09X + 0.59 | 0.21X + 0.56 |
| Chlorobenzene | 0.95C + 0.02 | 0.09X + 0.23 | 0.17X + 0.10 |
| 1,2-Dichlorobenzene | 0.93C + 0.52 | 0.17X −0.04 | 0.22X + 0.53 |
| 1,3-Dichlorobenzene | 0.96C−0.05 | 0.15X −0.10 | 0.19X + 0.09 |
| 1,4-Dichlorobenzene | 0.93C−0.09 | 0.15X + 0.28 | 0.20X + 0.41 |
| Ethylbenzene | 0.94C + 0.31 | 0.17X + 0.46 | 0.26X + 0.23 |
| Toluene | 0.94C + 0.65 | 0.09X + 0.48 | 0.18X + 0.71 |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
S′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in X µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the Concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
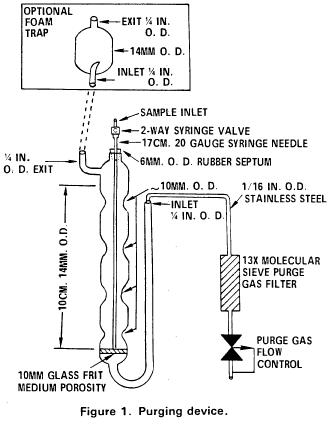
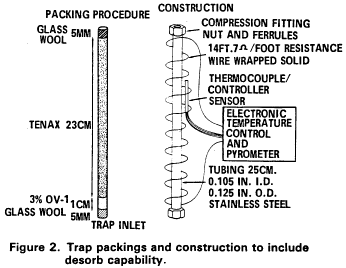
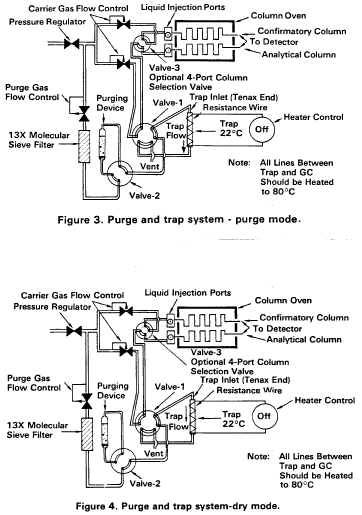
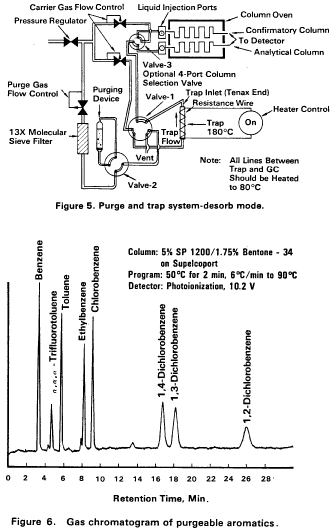 Method 603 -
Acrolein and Acrylonitrile 1. Scope and Application
Method 603 -
Acrolein and Acrylonitrile 1. Scope and Application
1.1 This method covers the determination of acrolein and acrylonitrile. The following parameters may be determined by this method:
| Parameter | STORET No. | CAS No. |
|---|---|---|
| Acrolein | 34210 | 107-02-8 |
| Acrylonitrile | 34215 | 107-13-1 |
1.2 This is a purge and trap gas chromatographic (GC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for either or both of the compounds above, compound identifications should be supported by at least one additional qualitative technique. This method describes analytical conditions for a second gas chromatographic column that can be used to confirm measurements made with the primary column. Method 624 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for the parameters listed above, if used with the purge and trap conditions described in this method.
1.3 The method detection limit (MDL, defined in Section 12.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.4 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.5 This method is restricted to use by or under the supervision of analysts experienced in the operation of a purge and trap system and a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 An inert gas is bubbled through a 5-mL water sample contained in a heated purging chamber. Acrolein and acrylonitrile are transferred from the aqueous phase to the vapor phase. The vapor is swept through a sorbent trap where the analytes are trapped. After the purge is completed, the trap is heated and backflushed with the inert gas to desorb the compound onto a gas chromatographic column. The gas chromatograph is temperature programmed to separate the analytes which are then detected with a flame ionization detector. 2 3
2.2 The method provides an optional gas chromatographic column that may be helpful in resolving the compounds of interest from the interferences that may occur.
3. Interferences3.1 Impurities in the purge gas and organic compound outgassing from the plumbing of the trap account for the majority of contamination problems. The analytical system must be demonstrated to be free from contamination under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3. The use of non-Teflon plastic tubing, non-Teflon thread sealants, or flow controllers with rubber components in the purge and trap system should be avoided.
3.2 Samples can be contaminated by diffusion of volatile organics through the septum seal into the sample during shipment and storage. A field reagent blank prepared from reagent water and carried through the sampling and handling protocol can serve as a check on such contamination.
3.3 Contamination by carry-over can occur whenever high level and low level samples are sequentially analyzed. To reduce carry-over, the purging device and sample syringe must be rinsed between samples with reagent water. Whenever an unusually concentrated sample is encountered, it should be followed by an analysis of reagent water to check for cross contamination. For samples containing large amounts of water-soluble materials, suspended solids, high boiling compounds or high analyte levels, it may be necessary to wash the purging device with a detergent solution, rinse it with distilled water, and then dry it in an oven at 105 °C between analyses. The trap and other parts of the system are also subject to contamination, therefore, frequent bakeout and purging of the entire system may be required.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this view point, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 4 6 for the information of the analyst.
5. Apparatus and Materials5.1 Sampling equipment, for discrete sampling.
5.1.1 Vial - 25-mL capacity or larger, equipped with a screw cap with a hole in the center (Pierce #13075 or equivalent). Detergent wash, rinse with tap and distilled water, and dry at 105 °C before use.
5.1.2 Septum - Teflon-faced silicone (Pierce #12722 or equivalent). Detergent wash, rinse with tap and distilled water and dry at 105 °C for 1 h before use.
5.2 Purge and trap system - The purge and trap system consists of three separate pieces of equipment: a purging device, trap, and desorber. Several complete systems are now commercially available.
5.2.1 The purging device must be designed to accept 5-mL, samples with a water column at least 3 cm deep. The gaseous head space between the water column and the trap must have a total volume of less than 15 mL. The purge gas must pass through the water column as finely divided bubbles with a diameter of less than 3 mm at the origin. The purge gas must be introduced no more than 5 mm from the base of the water column. The purging device must be capable of being heated to 85 °C within 3.0 min after transfer of the sample to the purging device and being held at 85 ±2 °C during the purge cycle. The entire water column in the purging device must be heated. Design of this modification to the standard purging device is optional, however, use of a water bath is suggested.
5.2.1.1 Heating mantle - To be used to heat water bath.
5.2.1.2 Temperature controller - Equipped with thermocouple/sensor to accurately control water bath temperature to ±2 °C. The purging device illustrated in Figure 1 meets these design criteria.
5.2.2 The trap must be at least 25 cm long and have an inside diameter of at least 0.105 in. The trap must be packed to contain 1.0 cm of methyl silicone coated packing (Section 6.5.2) and 23 cm of 2,6-diphenylene oxide polymer (Section 6.5.1). The minimum specifications for the trap are illustrated in Figure 2.
5.2.3 The desorber must be capable of rapidly heating the trap to 180 °C, The desorber illustrated in Figure 2 meets these design criteria.
5.2.4 The purge and trap system may be assembled as a separate unit as illustrated in Figure 3 or be coupled to a gas chromatograph.
5.3 pH paper - Narrow pH range, about 3.5 to 5.5 (Fisher Scientific Short Range Alkacid No. 2, #14-837-2 or equivalent).
5.4 Gas chromatograph - An analytical system complete with a temperature programmable gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.4.1 Column 1 - 10 ft long × 2 mm ID glass or stainless steel, packed with Porapak-QS (80/100 mesh) or equivalent. This column was used to develop the method performance statements in Section 12. Guidelines for the use of alternate column packings are provided in Section 10.1.
5.4.2 Column 2 - 6 ft long × 0.1 in. ID glass or stainless steel, packed with Chromosorb 101 (60/80 mesh) or equivalent.
5.4.3 Detector - Flame ionization detector. This type of detector has proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1), and was used to develop the method performance statements in Section 12. Guidelines for the use of alternate detectors are provided in Section 10.1.
5.5 Syringes - 5-mL, glass hypodermic with Luerlok tip (two each).
5.6 Micro syringes - 25-µL, 0.006 in. ID needle.
5.7 Syringe valve - 2-way, with Luer ends (three each).
5.8 Bottle - 15-mL, screw-cap, with Teflon cap liner.
5.9 Balance - Analytical, capable of accurately weighing 0.0001 g.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.1.1 Reagent water can be generated by passing tap water through a carbon filter bed containing about 1 lb of activated carbon (Filtrasorb-300, Calgon Corp., or equivalent).
6.1.2 A water purification system (Millipore Super-Q or equivalent) may be used to generate reagent water.
6.1.3 Regent water may also be prepared by boiling water for 15 min. Subsequently, while maintaining the temperature at 90 °C, bubble a contaminant-free inert gas through the water for 1 h. While still hot, transfer the water to a narrow mouth screw-cap bottle and seal with a Teflon-lined septum and cap.
6.2 Sodium thiosulfate - (ACS) Granular.
6.3 Sodium hydroxide solution (10 N) - Dissolve 40 g of NaOH (ACS) in reagent water and dilute to 100 mL.
6.4 Hydrochloric acid (1 + 1) - Slowly, add 50 mL of concentrated HCl (ACS) to 50 mL of reagent water.
6.5 Trap Materials:
6.5.1 2,6-Diphenylene oxide polymer - Tenax (60/80 mesh), chromatographic grade or equivalent.
6.5.2 Methyl silicone packing - 3% OV-1 on Chromosorb-W (60/80 mesh) or equivalent.
6.6 Stock standard solutions - Stock standard solutions may be prepared from pure standard materials or purchased as certified solutions. Prepare stock standard solutions in reagent water using assayed liquids. Since acrolein and acrylonitrile are lachrymators, primary dilutions of these compounds should be prepared in a hood. A NIOSH/MESA approved toxic gas respirator should be used when the analyst handles high concentrations of such materials.
6.6.1 Place about 9.8 mL of reagent water into a 10-mL ground glass stoppered volumetric flask. For acrolein standards the reagent water must be adjusted to pH 4 to 5. Weight the flask to the nearest 0.1 mg.
6.6.2 Using a 100-µL syringe, immediately add two or more drops of assayed reference material to the flask, then reweigh. Be sure that the drops fall directly into the water without contacting the neck of the flask.
6.6.3 Reweigh, dilute to volume, stopper, then mix by inverting the flask several times. Calculate the concentration in µg/µL from the net gain in weight. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock staldard. Optionally, stock standard solutions may be prepared using the pure standard material by volumetrically measuring the appropriate amounts and determining the weight of the material using the density of the material. Commercially prepared stock standards may be used at any concentration if they are certified by the manufactaurer or by an independent source.
6.6.4 Transfer the stock standard solution into a Teflon-sealed screw-cap bottle. Store at 4 °C and protect from light.
6.6.5 Prepare fresh standards daily.
6.7 Secondary dilution standards - Using stock standard solutions, prepare secondary dilution standards in reagent water that contain the compounds of interest, either singly or mixed together. The secondary dilution standards should be prepared at concentrations such that the aqueous calibration standards prepared in Section 7.3.1 or 7.4.1 will bracket the working range of the analytical system. Secondary dilution standards should be prepared daily and stored at 4 °C.
6.8 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Assemble a purge and trap system that meets the specifications in Section 5.2. Condition the trap overnight at 180 °C by backflushing with an inert gas flow of at least 20 mL/min. Condition the trap for 10 min once daily prior to use.
7.2 Connect the purge and trap system to a gas chromatograph. The gas chromatograph must be operated using temperature and flow rate conditions equivalent to those given in Table 1. Calibrate the purge and trap-gas chromatographic system using either the external standard technique (Section 7.3) or the internal standard technique (Section 7.4).
7.3 External standard calibration procedure:
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter by carefully adding 20.0 µL of one or more secondary dilution standards to 100, 500, or 1000 mL of reagent water. A 25-µL syringe with a 0.006 in. ID needle should be used for this operation. One of the external standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector. These standards must be prepared fresh daily.
7.3.2 Analyze each calibration standard according to Section 10, and tabulate peak height or area responses versus the concentration of the standard. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to concentration (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.4 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.4.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest as described in Section 7.3.1.
7.4.2 Prepare a spiking solution containing each of the internal standards using the procedures described in Sections 6.6 and 6.7. It is recommended that the secondary dilution standard be prepared at a concentration of 15 µg/mL of each internal standard compound. The addition of 10 µL of this standard to 5.0 mL of sample or calibration standard would be equivalent to 30 µg/L.
7.4.3 Analyze each calibration standard according to Section 10, adding 10 µL of internal standard spiking solution directly to the syringe (Section 10.4). Tabulate peak height or area responses against concentration for each compound and internal standard, and calculate response factors (RF) for each compound using Equation 1.
RF = (As)(Cis (Ais)(Cs) Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard. Cs = Concentration of the parameter to be measured. If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.7.5 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of a QC check sample.
7.5.1 Prepare the QC check sample as described in Section 8.2.2.
7.5.2 Analyze the QC check sample according to Section 10.
7.5.3 For each parameter, compare the response (Q) with the corresponding calibration acceptance criteria found in Table 2. If the responses for all parameters of interest fall within the designated ranges, analysis of actual samples can begin. If any individual Q falls outside the range, a new calibration curve, calibration factor, or RF must be prepared for that parameter according to Section 7.3 or 7.4.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Section 10.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Each day, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system are under control.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest at a concentration of 25 µg/mL in reagent water. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Prepare a QC check sample to contain 50 µg/L of each parameter by adding 200 µL of QC check sample concentrate to 100 mL of reagent water.
8.2.3 Analyze four 5-mL aliquots of the well-mixed QC check sample according to Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 3. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If either s exceeds the precision limit or X falls outside the range for accuracy, the system performance is unacceptable for that parameter. Locate and correct the source of the problem and repeat the test for each compound of interest.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at 50 µg/L or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.2 Analyze one 5-mL sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second 5-mL sample aliquot with 10 µL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100(A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 3. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 7
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 10 µL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 5 mL of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 3. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 All samples must be iced or refrigerated from the time of collection until analysis. If the sample contains free or combined chlorine, add sodium thiosulfate preservative (10 mg/40 mL is sufficient for up to 5 ppm Cl2) to the empty sample bottle just prior to shipping to the sampling site. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine. 8 Field test kits are available for this purpose.
9.2 If acrolein is to be analyzed, collect about 500 mL of sample in a clean glass container. Adjust the pH of the sample to 4 to 5 using acid or base, measuring with narrow range pH paper. Samples for acrolein analysis receiving no pH adjustment must be analyzed within 3 days of sampling.
9.3 Grab samples must be collected in glass containers having a total volume of at least 25 mL. Fill the sample bottle just to overflowing in such a manner that no air bubbles pass through the sample as the bottle is being filled. Seal the bottle so that no air bubbles are entrapped in it. If preservative has been added, shake vigorously for 1 min. Maintain the hermetic seal on the sample bottle until time of analysis.
9.4 All samples must be analyzed within 14 days of collection. 3
10. Procedure10.1 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are estimated retention times and MDL that can be achieved under these conditions. An example of the separations achieved by Column 1 is shown in Figure 5. Other packed columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
10.2 Calibrate the system daily as described in Section 7.
10.3 Adjust the purge gas (nitrogen or helium) flow rate to 20 mL-min. Attach the trap inlet to the purging device, and set the purge and trap system to purge (Figure 3). Open the syringe valve located on the purging device sample introduction needle.
10.4 Remove the plunger from a 5-mL syringe and attach a closed syringe valve. Open the sample bottle (or standard) and carefully pour the sample into the syringe barrel to just short of overflowing. Replace the syringe plunger and compress the sample. Open the syringe valve and vent any residual air while adjusting the sample volume to 5.0 mL. Since this process of taking an aliquot destroys the validity of the sample for future analysis, the analyst should fill a second syringe at this time to protect against possible loss of data. Add 10.0 µL of the internal standard spiking solution (Section 7.4.2), if applicable, through the valve bore then close the valve.
10.5 Attach the syringe-syringe valve assembly to the syringe valve on the purging device. Open the syringe valves and inject the sample into the purging chamber.
10.6 Close both valves and purge the sample for 15.0 ±0.1 min while heating at 85 ±2 °C.
10.7 After the 15-min purge time, attach the trap to the chromatograph, adjust the purge and trap system to the desorb mode (Figure 4), and begin to temperature program the gas chromatograph. Introduce the trapped materials to the GC column by rapidly heating the trap to 180 °C while backflushing the trap with an inert gas between 20 and 60 mL/min for 1.5 min.
10.8 While the trap is being desorbed into the gas chromatograph, empty the purging chamber using the sample introduction syringe. Wash the chamber with two 5-mL flushes of reagent water.
10.9 After desorbing the sample for 1.5 min, recondition the trap by returning the purge and trap system to the purge mode. Wait 15 s then close the syringe valve on the purging device to begin gas flow through the trap. The trap temperature should be maintained at 210 °C. After approximately 7 min, turn off the trap heater and open the syringe valve to stop the gas flow through the trap. When the trap is cool, the next sample can be analyzed.
10.10 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
11. Calculations11.1 Determine the concentration of individual compounds in the sample.
11.1.1 If the external standard calibration procedure is used, calculate the concentration of the parameter being measured from the peak response using the calibration curve or calibration factor determined in Section 7.3.2.
11.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.4.3 and Equation 2.
Equation 2 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard.11.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
12. Method Performance12.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Table 1 were obtained using reagent water. 9 The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
12.2 This method is recommended for the concentration range from the MDL to 1,000 × MDL. Direct aqueous injection techniques should be used to measure concentration levels above 1,000 × MDL.
12.3 In a single laboratory (Battelle-Columbus), the average recoveries and standard deviations presented in Table 2 were obtained. 9 Seven replicate samples were analyzed at each spike level.
References1. 40 CFR part 136, appendix B.
2. Bellar, T.A., and Lichtenberg, J.J. “Determining Volatile Organics at Microgram-per-Litre-Levels by Gas Chromatography,” Journal American Water Works Association, 66, 739 (1974).
3. “Evaluate Test Procedures for Acrolein and Acrylonitrile,” Special letter report for EPA Project 4719-A, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, 27 June 1979.
4. “Carcinogens - Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
5. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
7. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983).
8. “Methods 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric, DPD) for Chlorine, Total Residual,” Methods for Chemical Analysis of Water and Wastes, EPA-600/4-79-020, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1979.
9. “Evaluation of Method 603 (Modified),” EPA-600/4-84-ABC, National Technical Information Service, PB84-, Springfield, Virginia 22161, Nov. 1984.
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Method detection limit (µg/L) | |
|---|---|---|---|
| Column 1 | Column 2 | ||
| Acrolein | 10.6 | 8.2 | 0.7 |
| Acrylonitrile | 12.7 | 9.8 | 0.5 |
Column 1 conditions: Porapak-QS (80/100 mesh) packed in a 10 ft × 2 mm ID glass or stainless steel column with helium carrier gas at 30 mL/min flow rate. Column temperature held isothermal at 110 °C for 1.5 min (during desorption), then heated as rapidly as possible to 150 °C and held for 20 min; column bakeout at 190 °C for 10 min. 9
Column 2 conditions: Chromosorb 101 (60/80 mesh) packed in a 6 ft. × 0.1 in. ID glass or stainless steel column with helium carrier gas at 40 mL/min flow rate. Column temperature held isothermal at 80 °C for 4 min, then programmed at 50 °C/min to 120 °C and held for 12 min.
Table 2 - Single Laboratory Accuracy and Precision - Method 603
| Parameter | Sample matrix | Spike conc. (µg/L) | Average recovery (µg/L) | Standard deviation (µg/L) | Average percent recovery |
|---|---|---|---|---|---|
| Acrolein | RW | 5.0 | 5.2 | 0.2 | 104 |
| RW | 50.0 | 51.4 | 0.7 | 103 | |
| POTW | 5.0 | 4.0 | 0.2 | 80 | |
| POTW | 50.0 | 44.4 | 0.8 | 89 | |
| IW | 5.0 | 0.1 | 0.1 | 2 | |
| IW | 100.0 | 9.3 | 1.1 | 9 | |
| Acrylonitrile | RW | 5.0 | 4.2 | 0.2 | 84 |
| RW | 50.0 | 51.4 | 1.5 | 103 | |
| POTW | 20.0 | 20.1 | 0.8 | 100 | |
| POTW | 100.0 | 101.3 | 1.5 | 101 | |
| IW | 10.0 | 9.1 | 0.8 | 91 | |
| IW | 100.0 | 104.0 | 3.2 | 104 |
RW = Reagent water.
POTW = Prechlorination secondary effluent from a municipal sewage treatment plant.
IW = Industrial wastewater containing an unidentified acrolein reactant.
Table 3 - Calibration and QC Acceptance Criteria - Method 603 a
| Parameter | Range for Q (µg/L) | Limit for S (µg/L) | Range for X (µg/L) | Range for P, Ps (%) |
|---|---|---|---|---|
| Acrolein | 45.9-54.1 | 4.6 | 42.9-60.1 | 88-118 |
| Acrylonitrile | 41.2-58.8 | 9.9 | 33.1-69.9 | 71-135 |
a = Criteria were calculated assuming a QC check sample concentration of 50 µg/L. 9
Q = Concentration measured in QC check sample, in µg/L (Section 7.5.3).
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
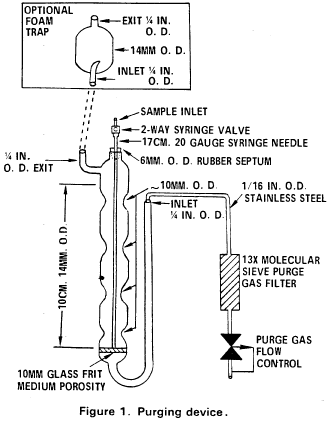
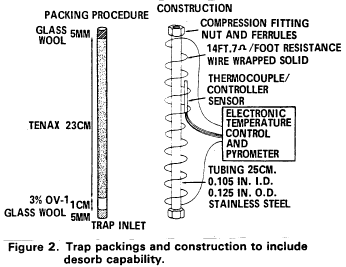

 Method 604 -
Phenols 1. Scope and Application
Method 604 -
Phenols 1. Scope and Application
1.1 This method covers the determination of phenol and certain substituted phenols. The following parameters may be determined by this method:
| Parameter | STORET No. | CAS No. |
|---|---|---|
| 4-Chloro-3-methylphenol | 34452 | 59-50-7 |
| 2--Chlorophenol | 34586 | 95-57-8 |
| 2,4-Dichlorophenol | 34601 | 120-83-2 |
| 2,4-Dimethylphenol | 34606 | 105-67-9 |
| 2,4-Dinitrophenol | 34616 | 51-28-5 |
| 2-Methyl-4,6-dinitrophenol | 34657 | 534-52-1 |
| 2-Nitrophenol | 34591 | 88-75-5 |
| 4-Nitrophenol | 34646 | 100-02-7 |
| Pentachlorophenol | 39032 | 87-86-5 |
| Phenol | 34694 | 108-95-2 |
| 2,4,6-Trichlorophenol | 34621 | 88-06-2 |
1.2 This is a flame ionization detector gas chromatographic (FIDGC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compounds above, compound identifications should be supported by at least one additional qualitative technique. This method describes analytical conditions for derivatization, cleanup, and electron capture detector gas chromatography (ECDGC) that can be used to confirm measurements made by FIDGC. Method 625 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for all of the parameters listed above, using the extract produced by this method.
1.3 The method detection limit (MDL, defined in Section 14.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix. The MDL listed in Table 1 for each parameter was achieved with a flame ionization detector (FID). The MDLs that were achieved when the derivatization cleanup and electron capture detector (ECD) were employed are presented in Table 2.
1.4 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.5 This method is restricted to use by or under the supervision of analysts experienced in the use of a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is acidified and extracted with methylene chloride using a separatory funnel. The methylene chloride extract is dried and exchanged to 2-propanol during concentration to a volume of 10 mL or less. The extract is separated by gas chromatography and the phenols are then measured with an FID. 2
2.2 A preliminary sample wash under basic conditions can be employed for samples having high general organic and organic base interferences.
2.3 The method also provides for a derivatization and column chromatography cleanup procedure to aid in the elimination of interferences. 2 3 The derivatives are analyzed by ECDGC.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardware that lead to discrete artifacts and/or elevated baselines in gas chromatograms. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 4 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Some thermally stable materials, such as PCBs, may not be eliminated by this treatment. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Thorough rinsing with such solvents usually eliminates PCB interference. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to minimize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Matrix interferences may be caused by contaminants that are coextracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. The derivatization cleanup procedure in Section 12 can be used to overcome many of these interferences, but unique samples may require additional cleanup approaches to achieve the MDL listed in Tables 1 and 2.
3.3 The basic sample wash (Section 10.2) may cause significantly reduced recovery of phenol and 2,4-dimethylphenol. The analyst must recognize that results obtained under these conditions are minimum concentrations.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this mothod has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 5 7 for the information of analyst.
4.2 Special care should be taken in handling pentafluorobenzyl bromide, which is a lachrymator, and 18-crown-6-ether, which is highly toxic.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1-L or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flow meter is required to collect flow proportional composites.
5.2 Glassware (All specifications are suggested. Catalog numbers are included for illustration only.):
5.2.1 Separatory funnel - 2-L, with Teflon stopcock.
5.2.2 Drying column - Chromatographic column, 400 mm long × 19 mm ID, with coarse frit filter disc.
5.2.3 Chromatographic column - 100 mm long × 10 mm ID, with Teflon stopcock.
5.2.4 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes K-570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. Ground glass stopper is used to prevent evaporation of extracts.
5.2.5 Evaporative flask, Kuderna-Danish - 500-mL (Kontes K-570001-0500 or equivalent). Attach to concentrator tube with springs.
5.2.6 Snyder column, Kuderna-Danish - Three-ball macro (Kontes K-503000-0121 or equivalent).
5.2.7 Snyder column, Kuderna-Danish - Two-ball micro (Kontes K-569001-0219 or equivalent).
5.2.8 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.2.9 Reaction flask - 15 to 25-mL round bottom flask, with standard tapered joint, fitted with a water-cooled condenser and U-shaped drying tube containing granular calcium chloride.
5.3 Boiling chips - Approximately 10/40 mesh. Heat to 400 °C for 30 min or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 Balance - Analytical, capable of accurately weighting 0.0001 g.
5.6 Gas chromatograph - An analytical system complete with a temperature programmable gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.6.1 Column for underivatized phenols - 1.8 m long × 2 mm ID glass, packed with 1% SP-1240DA on Supelcoport (80/100 mesh) or equivalent. This column was used to develop the method performance statements in Section 14. Guidelines for the use of alternate column packings are provided in Section 11.1.
5.6.2 Column for derivatized phenols - 1.8 m long × 2 mm ID glass, packed with 5% OV-17 on Chromosorb W-AW-DMCS (80/100 mesh) or equivalent. This column has proven effective in the analysis of wastewaters for derivatization products of the parameters listed in the scope (Section 1.1), and was used to develop the method performance statements in Section 14. Guidelines for the use of alternate column packings are provided in Section 11.1.
5.6.3 Detectors - Flame ionization and electron capture detectors. The FID is used when determining the parent phenols. The ECD is used when determining the derivatized phenols. Guidelines for the use of alternatve detectors are provided in Section 11.1.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.2 Sodium hydroxide solution (10 N) - Dissolve 40 g of NaOH (ACS) in reagent water and dilute to 100 mL.
6.3 Sodium hydroxide solution (1 N) - Dissolve 4 g of NaOH (ACS) in reagent water and dilute to 100 mL.
6.4 Sodium sulfate - (ACS) Granular, anhydrous. Purify by heating at 400 °C for 4 h in a shallow tray.
6.5 Sodium thiosulfate - (ACS) Granular.
6.6 Sulfuric acid (1 + 1) - Slowly, add 50 mL of H2SO4 (ACS, sp. gr. 1.84) to 50 mL of reagent water.
6.7 Sulfuric acid (1 N) - Slowly, add 58 mL of H2SO4 (ACS, sp. gr. 1.84) to reagent water and dilute to 1 L.
6.8 Potassium carbonate - (ACS) Powdered.
6.9 Pentafluorobenzyl bromide (α-Bromopentafluorotoluene) - 97% minimum purity.
Note:This chemical is a lachrymator. (See Section 4.2.)
6.10 18-crown-6-ether (1,4,7,10,13,16-Hexaoxacyclooctadecane) - 98% minimum purity.
Note:This chemical is highly toxic.
6.11 Derivatization reagent - Add 1 mL of pentafluorobenzyl bromide and 1 g of 18-crown-6-ether to a 50-mL volumetric flask and dilute to volume with 2-propanol. Prepare fresh weekly. This operation should be carried out in a hood. Store at 4 °C and protect from light.
6.12 Acetone, hexane, methanol, methylene chloride, 2-propanol, toluene - Pesticide quality or equivalent.
6.13 Silica gel - 100/200 mesh, Davison, grade-923 or equivalent. Activate at 130 °C overnight and store in a desiccator.
6.14 Stock standard solutions (1.00 µg/µL) - Stock standard solutions may be prepared from pure standard materials or purchased as certified solutions.
6.14.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in 2-propanol and dilute to volume in a 10-mL volumetric flask. Larger volumes can be used at the convenience of the analyst. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.14.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store at 4 °C and protect from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.14.3 Stock standard solutions must be replaced after six months, or sooner if comparison with check standards indicates a problem.
6.15 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 To calibrate the FIDGC for the anaylsis of underivatized phenols, establish gas chromatographic operating conditions equivalent to those given in Table 1. The gas chromatographic system can be calibrated using the external standard technique (Section 7.2) or the internal standard technique (Section 7.3).
7.2 External standard calibration procedure for FIDGC:
7.2.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with 2-propanol. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.2.2 Using injections of 2 to 5 µl, analyze each calibration standard according to Section 11 and tabulate peak height or area responses against the mass injected. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to amount injected (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.3 Internal standard calibration procedure for FIDGC - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask. To each calibration standard, add a known constant amount of one or more internal standards, and dilute to volume with 2-propanol. One of the standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.3.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 11 and tabulate peak height or area responses against concentration for each compound and internal standard. Calculate response factors (RF) for each compound using Equation 1.
RF = (As)(Cis (Ais)(Cs) Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of the parameter to be measured (µg/L).If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.
7.4 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of one or more calibration standards. If the response for any parameter varies from the predicted response by more than ±15%, a new calibration curve must be prepared for that compound.
7.5 To calibrate the ECDGC for the analysis of phenol derivatives, establish gas chromatographic operating conditions equivalent to those given in Table 2.
7.5.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with 2-propanol. One of the external standards should be at a concentration near, but above, the MDL (Table 2) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.5.2 Each time samples are to be derivatized, simultaneously treat a 1-mL aliquot of each calibration standard as described in Section 12.
7.5.3 After derivatization, analyze 2 to 5 µL of each column eluate collected according to the method beginning in Section 12.8 and tabulate peak height or area responses against the calculated equivalent mass of underivatized phenol injected. The results can be used to prepare a calibration curve for each compound.
7.6 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.6 and 11.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Before processing any samples the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest at a concentration of 100 µg/mL in 2-propanol. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at a concentration of 100 µg/L by adding 1.00 mL of QC check sample concentrate to each of four 1-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 3. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter.
Note:The large number of parameters in Talbe 3 present a substantial probability that one or more will fail at least one of the acceptance criteria when all parameters are analyzed.
8.2.6 When one or more of the parameters tested fail at least one of the acceptance criteria, the analyst must proceed according to Section 8.2.6.1 or 8.2.6.2.
8.2.6.1 Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.2.
8.2.6.2 Beginning with Section 8.2.2, repeat the test only for those parameters that failed to meet criteria. Repeated failure, however, will confirm a general problem with the measurement system. If this occurs, locate and correct the source of the problem and repeat the test for all compounds of interest beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at 100 µg/L or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determine background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any, or, if none, (2) the larger of either 5 times higher than the expected background concentration or 100 µg/L.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100(A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 3. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 8 If spiking was performed at a concentration lower than 100 µg/L, the analyst must use either the QC acceptance criteria in Table 3, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 4, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 4, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T)±2.44(100 S′/T)%. 8
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 1.0 mL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 1 L of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 3. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6. It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 9 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C from the time of collection until extraction. Fill the sample bottles and, if residual chlorine is present, add 80 mg of sodium thiosulfate per liter of sample and mix well. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine. 10 Field test kits are available for this purpose.
9.3 All samples must be extracted within 7 days of collection and completely analyzed within 40 days of extraction. 2
10. Sample Extraction10.1 Mark the water meniscus on the side of sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel.
10.2 For samples high in organic content, the analyst may solvent wash the sample at basic pH as prescribed in Sections 10.2.1 and 10.2.2 to remove potential method interferences. Prolonged or exhaustive contact with solvent during the wash may result in low recovery of some of the phenols, notably phenol and 2,4-dimethylphenol. For relatively clean samples, the wash should be omitted and the extraction, beginning with Section 10.3, should be followed.
10.2.1 Adjust the pH of the sample to 12.0 or greater with sodium hydroxide solution.
10.2.2 Add 60 mL of methylene chloride to the sample by shaking the funnel for 1 min with periodic venting to release excess pressure. Discard the solvent layer. The wash can be repeated up to two additional times if significant color is being removed.
10.3 Adjust the sample to a pH of 1 to 2 with sulfuric acid.
10.4 Add 60 mL of methylene chloride to the sample bottle, seal, and shake 30 s to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min. with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the methylene chloride extract in a 250-mL Erlenmeyer flask.
10.5 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.6 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator if the requirements of Section 8.2 are met.
10.7 Pour the combined extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the Erlenmeyer flask and column with 20 to 30 mL of methylene chloride to complete the quantitative transfer.
10.8 Add one or two clean boiling chips to the evaporative flask and attach a three-ball Snyder column. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
10.9 Increase the temperature of the hot water bath to 95 to 100 °C. Remove the Synder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of 2-propanol. A 5-mL syringe is recommended for this operation. Attach a two-ball micro-Snyder column to the concentrator tube and prewet the column by adding about 0.5 mL of 2-propanol to the top. Place the micro-K-D apparatus on the water bath so that the concentrator tube is partially immersed in the hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete concentration in 5 to 10 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood. When the apparent volume of liquid reaches 2.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min. Add an additional 2 mL of 2-propanol through the top of the micro-Snyder column and resume concentrating as before. When the apparent volume of liquid reaches 0.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
10.10 Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with a minimum amount of 2-propanol. Adjust the extract volume to 1.0 mL. Stopper the concentrator tube and store refrigerated at 4 °C if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial. If the sample extract requires no further cleanup, proceed with FIDGC analysis (Section 11). If the sample requires further cleanup, proceed to Section 12.
10.11 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1000-mL graduated cylinder. Record the sample volume to the nearest 5 mL.
11. Flame Ionization Detector Gas Chromatography11.1 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and MDL that can be achieved under these conditions. An example of the separations achieved by this column is shown in Figure 1. Other packed or capillary (open-tubular) columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
11.2 Calibrate the system daily as described in Section 7.
11.3 If the internal standard calibration procedure is used, the internal standard must be added to the sample extract and mixed thoroughly immediately before injection into the gas chromatograph.
11.4 Inject 2 to 5 µL of the sample extract or standard into the gas chromatograph using the solvent-flush technique. 11 Smaller (1.0 µL) volumes may be injected if automatic devices are employed. Record the volume injected to the nearest 0.05 µL, and the resulting peak size in area or peak height units.
11.5 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound may be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
11.6 If the response for a peak exceeds the working range of the system, dilute the extract and reanalyze.
11.7 If the measurement of the peak response is prevented by the presence of interferences, an alternative gas chromatographic procedure is required. Section 12 describes a derivatization and column chromatographic procedure which has been tested and found to be a practical means of analyzing phenols in complex extracts.
12. Derivatization and Electron Capture Detector Gas Chromatography12.1 Pipet a 1.0-mL aliquot of the 2-propanol solution of standard or sample extract into a glass reaction vial. Add 1.0 mL of derivatizing reagent (Section 6.11). This amount of reagent is sufficient to derivatize a solution whose total phenolic content does not exceed 0.3 mg/mL.
12.2 Add about 3 mg of potassium carbonate to the solution and shake gently.
12.3 Cap the mixture and heat it for 4 h at 80 °C in a hot water bath.
12.4 Remove the solution from the hot water bath and allow it to cool.
12.5 Add 10 mL of hexane to the reaction flask and shake vigorously for 1 min. Add 3.0 mL of distilled, deionized water to the reaction flask and shake for 2 min. Decant a portion of the organic layer into a concentrator tube and cap with a glass stopper.
12.6 Place 4.0 g of silica gel into a chromatographic column. Tap the column to settle the silica gel and add about 2 g of anhydrous sodium sulfate to the top.
12.7 Preelute the column with 6 mL of hexane. Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, pipet onto the column 2.0 mL of the hexane solution (Section 12.5) that contains the derivatized sample or standard. Elute the column with 10.0 mL of hexane and discard the eluate. Elute the column, in order, with: 10.0 mL of 15% toluene in hexane (Fraction 1); 10.0 mL of 40% toluene in hexane (Fraction 2); 10.0 mL of 75% toluene in hexane (Fraction 3); and 10.0 mL of 15% 2-propanol in toluene (Fraction 4). All elution mixtures are prepared on a volume: volume basis. Elution patterns for the phenolic derivatives are shown in Table 2. Fractions may be combined as desired, depending upon the specific phenols of interest or level of interferences.
12.8 Analyze the fractions by ECDGC. Table 2 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and MDL that can be achieved under these conditions. An example of the separations achieved by this column is shown in Figure 2.
12.9 Calibrate the system daily with a minimum of three aliquots of calibration standards, containing each of the phenols of interest that are derivatized according to Section 7.5.
12.10 Inject 2 to 5 µL of the column fractions into the gas chromatograph using the solvent-flush technique. Smaller (1.0 µL) volumes can be injected if automatic devices are employed. Record the volume injected to the nearest 0.05 µL, and the resulting peak size in area or peak height units. If the peak response exceeds the linear range of the system, dilute the extract and reanalyze.
13. Calculations13.1 Determine the concentration of individual compounds in the sample analyzed by FIDGC (without derivatization) as indicated below.
13.1.1 If the external standard calibration procedure is used, calculate the amount of material injected from the peak response using the calibration curve or calibration factor determined in Section 7.2.2. The concentration in the sample can be calculated from Equation 2.
Equation 2 where: A = Amount of material injected (ng). Vi = Volume of extract injected (µL). Vt = Volume of total extract (µL). Vs = Volume of water extracted (mL).13.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.3.2 and Equation 3.
Equation 3 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).13.2 Determine the concentration of individual compounds in the sample analyzed by derivatization and ECDGC according to Equation 4.
Equation 4 where: A = Mass of underivatized phenol represented by area of peak in sample chromatogram, determined from calibration curve in Section 7.5.3 (ng). Vi = Volume of eluate injected (µL). Vt = Total volume of column eluate or combined fractions from which Vi was taken (µL). Vs = Volume of water extracted in Section 10.10 (mL). B = Total volume of hexane added in Section 12.5 (mL). C = Volume of hexane sample solution added to cleanup column in Section 12.7 (mL). D = Total volume of 2-propanol extract prior to derivatization (mL). E = Volume of 2-propanol extract carried through derivatization in Section 12.1 (mL).13.3 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
14. Method Performance14.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Tables 1 and 2 were obtained using reagent water. 12 Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
14.2 This method was tested by 20 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked as six concentrations over the range 12 to 450 µg/L. 13 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships for a flame ionization detector are presented in Table 4.
References1. 40 CFR part 136, appendix B.
2. “Determination of Phenols in Industrial and Municipal Wastewaters,” EPA 600/4-84-ABC, National Technical Information Service, PBXYZ, Springfield, Virginia 22161, November 1984.
3. Kawahara, F. K. “Microdetermination of Derivatives of Phenols and Mercaptans by Means of Electron Capture Gas Chromatography,” Analytical Chemistry, 40, 1009 (1968).
4. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia.
5. “Carcinogens - Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
6. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
7. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
8. Provost, L. P., and Elder, R. S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
9. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
10. “Methods 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric, DPD) for Chlorine, Total Residual,” Methmds for Chemical Analysis of Water and Wastes, EPA-600/4-79-020, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1979.
11. Burke, J. A. “Gas Chromatography for Pesticide Residue Analysis; Some Practical Aspects,” Journal of the Association of Official Analytical Chemists, 48, 1037 (1965).
12. “Development of Detection Limits, EPA Method 604, Phenols,” Special letter report for EPA Contract 68-03-2625, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268.
13. “EPA Method Study 14 Method 604-Phenols,” EPA 600/4-84-044, National Technical Information Service, PB84-196211, Springfield, Virginia 22161, May 1984.
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Method detection limit (µg/L) |
|---|---|---|
| 2-Chlorophenol | 1.70 | 0.31 |
| 2-Nitrophenol | 2.00 | 0.45 |
| Phenol | 3.01 | 0.14 |
| 2,4-Dimethylphenol | 4.03 | 0.32 |
| 2,4-Dichlorophenol | 4.30 | 0.39 |
| 2,4,6-Trichlorophenol | 6.05 | 0.64 |
| 4-Chloro-3-methylphenol | 7.50 | 0.36 |
| 2,4-Dinitrophenol | 10.00 | 13.0 |
| 2-Methyl-4,6-dinitrophenol | 10.24 | 16.0 |
| Pentachlorophenol | 12.42 | 7.4 |
| 4-Nitrophenol | 24.25 | 2.8 |
Column conditions: Supelcoport (80/100 mesh) coated with 1% SP-1240DA packed in a 1.8 m long × 2 mm ID glass column with nitrogen carrier gas at 30 mL/min flow rate. Column temperature was 80 °C at injection, programmed immediately at 8 °C/min to 150 °C final temperature. MDL were determined with an FID.
Table 2 - Silica Gel Fractionation and Electron Capture Gas Chromatography of PFBB Derivatives
| Parent compound | Percent recovery by fraction a | Retention time (min) | Method detection limit (µg/L) | |||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| 2-Chlorophenol | 90 | 1 | 3.3 | 0.58 | ||
| 2-Nitrophenol | 9 | 90 | 9.1 | 0.77 | ||
| Phenol | 90 | 10 | 1.8 | 2.2 | ||
| 2,4-Dimethylphenol | 95 | 7 | 2.9 | 0.63 | ||
| 2,4-Dichlorophenol | 95 | 1 | 5.8 | 0.68 | ||
| 2,4,6-Trichlorophenol | 50 | 50 | 7.0 | 0.58 | ||
| 4-Chloro-3-methylphenol | 84 | 14 | 4.8 | 1.8 | ||
| Pentachlorophenol | 75 | 20 | 28.8 | 0.59 | ||
| 4-Nitrophenol | 1 | 90 | 14.0 | 0.70 | ||
Column conditions: Chromosorb W-AW-DMCS (80/100 mesh) coated with 5% OV-17 packed in a 1.8 m long × 2.0 mm ID glass column with 5% methane/95% argon carrier gas at 30 mL/min flow rate. Column temperature held isothermal at 200 °C. MDL were determined with an ECD.
a Eluant composition:
Fraction 1 - 15% toluene in hexane.
Fraction 2 - 40% toluene in hexane.
Fraction 3 - 75% toluene in hexane.
Fraction 4 - 15% 2-propanol in toluene.
Table 3 - QC Acceptance Criteria - Method 604
| Parameter | Test conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps (percent) |
|---|---|---|---|---|
| 4-Chloro-3-methylphenol | 100 | 16.6 | 56.7-113.4 | 49-122 |
| 2-Chlorophenol | 100 | 27.0 | 54.1-110.2 | 38-126 |
| 2,4-Dichlorophenol | 100 | 25.1 | 59.7-103.3 | 44-119 |
| 2,4-Dimethylphenol | 100 | 33.3 | 50.4-100.0 | 24-118 |
| 4,6-Dinitro-2-methylphenol | 100 | 25.0 | 42.4-123.6 | 30-136 |
| 2,4-Dinitrophenol | 100 | 36.0 | 31.7-125.1 | 12-145 |
| 2-Nitrophenol | 100 | 22.5 | 56.6-103.8 | 43-117 |
| 4-Nitrophenol | 100 | 19.0 | 22.7-100.0 | 13-110 |
| Pentachlorophenol | 100 | 32.4 | 56.7-113.5 | 36-134 |
| Phenol | 100 | 14.1 | 32.4-100.0 | 23-108 |
| 2,4,6-Trichlorophenol | 100 | 16.6 | 60.8-110.4 | 53-119 |
s - Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X - Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps - Percent recovery measured (Section 8.3.2, Section 8.4.2).
Note: These criteria are based directly upon the method performance data in Table 4. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 4.
Table 4 - Method Accuracy and Precision as Functions of Concentration - Method 604
| Parameter | Accuracy, as recovery, X′ (µg/L) | Single Analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| 4-Chloro-3-methylphenol | 0.87C-1.97 | 0.11X -0.21 | 0.16X + 1.41 |
| 2-Chlorophenol | 0.83C-0.84 | 0.18X + 0.20 | 0.21X + 0.75 |
| 2,4-Dichlorophenol | 0.81C + 0.48 | 0.17X -0.02 | 0.18X + 0.62 |
| 2,4-Dimethylphenol | 0.62C-1.64 | 0.30X -0.89 | 0.25X + 0.48 |
| 4,6-Dinitro-2-methylphenol | 0.84C-1.01 | 0.15X + 1.25 | 0.19X + 5.85 |
| 2,4-Dinitrophenol | 0.80C-1.58 | 0.27X -1.15 | 0.29X + 4.51 |
| 2-Nitrophenol | 0.81C-0.76 | 0.15X + 0.44 | 0.14X + 3.84 |
| 4-Nitrophenol | 0.46C + 0.18 | 0.17X + 2.43 | 0.19X + 4.79 |
| Pentachlorophenol | 0.83C + 2.07 | 0.22X -0.58 | 0.23X + 0.57 |
| Phenol | 0.43C + 0.11 | 0.20X -0.88 | 0.17X + 0.77 |
| 2,4,6-Trichlorophenol | 0.86C-0.40 | 0.10X + 0.53 | 0.13X + 2.40 |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
 Method 605 -
Benzidines 1. Scope and Application
Method 605 -
Benzidines 1. Scope and Application
1.1 This method covers the determination of certain benzidines. The following parameters can be determined by this method:
| Parameter | Storet No | CAS No. |
|---|---|---|
| Benzidine | 39120 | 92-87-5 |
| 3,3′-Dichlorobenzidine | 34631 | 91-94-1 |
1.2 This is a high performance liquid chromatography (HPLC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for the compounds above, identifications should be supported by at least one additional qualitative technique. This method describes electrochemical conditions at a second potential which can be used to confirm measurements made with this method. Method 625 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for the parameters listed above, using the extract produced by this method.
1.3 The method detection limit (MDL, defined in Section 14.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of the interferences in the sample matrix.
1.4 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.5 This method is restricted to use by or under the supervision of analysts experienced in the use of HPLC instrumentation and in the interpretation of liquid chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is extracted with chloroform using liquid-liquid extractions in a separatory funnel. The chloroform extract is extracted with acid. The acid extract is then neutralized and extracted with chloroform. The final chloroform extract is exchanged to methanol while being concentrated using a rotary evaporator. The extract is mixed with buffer and separated by HPLC. The benzidine compounds are measured with an electrochemical detector. 2
2.2 The acid back-extraction acts as a general purpose cleanup to aid in the elimination of interferences.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardware that lead to discrete artifacts and/or elevated baselines in chromatograms. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 3 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Some thermally stable materials may not be eliminated by this treatment. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to minimize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Matrix interferences may be caused by contaminants that are co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. The cleanup procedures that are inherent in the extraction step are used to overcome many of these interferences, but unique samples may require additional cleanup approaches to achieve the MDL listed in Table 1.
3.3 Some dye plant effluents contain large amounts of components with retention times closed to benzidine. In these cases, it has been found useful to reduce the electrode potential in order to eliminate interferences and still detect benzidine. (See Section 12.7.)
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health harzard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 4 6 for the information of the analyst.
4.2 The following parameters covered by this method have been tentatively classified as known or suspected, human or mammalian carcinogens: benzidine and 3,3′-dichlorobenzidine. Primary standards of these toxic compounds should be prepared in a hood. A NIOSH/MESA approved toxic gas respirator should be worn when the analyst handles high concentrations of these toxic compounds.
4.3 Exposure to chloroform should be minimized by performing all extractions and extract concentrations in a hood or other well-ventiliated area.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1-L or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flow meter is required to collect flow proportional composites.
5.2 Glassware (All specifications are suggested):
5.2.1 Separatory funnels - 2000, 1000, and 250-mL, with Teflon stopcock.
5.2.2 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.2.3 Rotary evaporator.
5.2.4 Flasks - Round bottom, 100-mL, with 24/40 joints.
5.2.5 Centrifuge tubes - Conical, graduated, with Teflon-lined screw caps.
5.2.6 Pipettes - Pasteur, with bulbs.
5.3 Balance - Analytical, capable of accurately weighing 0.0001 g.
5.4 High performance liquid chromatograph (HPLC) - An analytical system complete with column supplies, high pressure syringes, detector, and compatible recorder. A data system is recommended for measuring peak areas and retention times.
5.4.1 Solvent delivery system - With pulse damper, Altex 110A or equivalent.
5.4.2 Injection valve (optional) - Waters U6K or equivalent.
5.4.3 Electrochemical detector - Bioanalytical Systems LC-2A with glassy carbon electrode, or equivalent. This detector has proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1), and was used to develop the method performance statements in Section 14. Guidelines for the use of alternate detectors are provided in Section 12.1.
5.4.4 Electrode polishing kit - Princeton Applied Research Model 9320 or equivalent.
5.4.5 Column - Lichrosorb RP-2, 5 micron particle diameter, in a 25 cm × 4.6 mm ID stainless steel column. This column was used to develop the method performance statements in Section 14. Guidelines for the use of alternate column packings are provided in Section 12.1.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.2 Sodium hydroxide solution (5 N) - Dissolve 20 g of NaOH (ACS) in reagent water and dilute to 100 mL.
6.3 Sodium hydroxide solution (1 M) - Dissolve 40 g of NaOH (ACS) in reagent water and dilute to 1 L.
6.4 Sodium thiosulfate - (ACS) Granular.
6.5 Sodium tribasic phosphate (0.4 M) - Dissolve 160 g of trisodium phosphate decahydrate (ACS) in reagent water and dilute to 1 L.
6.6 Sulfuric acid (1 + 1) - Slowly, add 50 mL of H2SO4 (ACS, sp. gr. 1.84) to 50 mL of reagent water.
6.7 Sulfuric acid (1 M) - Slowly, add 58 mL of H2SO4 (ACS, sp. gr. 1.84) to reagent water and dilute to 1 L.
6.8 Acetate buffer (0.1 M, pH 4.7) - Dissolve 5.8 mL of glacial acetic acid (ACS) and 13.6 g of sodium acetate trihydrate (ACS) in reagent water which has been purified by filtration through a RO-4 Millipore System or equivalent and dilute to 1 L.
6.9 Acetonitrile, chloroform (preserved with 1% ethanol), methanol - Pesticide quality or equivalent.
6.10 Mobile phase - Place equal volumes of filtered acetonitrile (Millipore type FH filter or equivalent) and filtered acetate buffer (Millipore type GS filter or equivalent) in a narrow-mouth, glass container and mix thoroughly. Prepare fresh weekly. Degas daily by sonicating under vacuum, by heating and stirring, or by purging with helium.
6.11 Stock standard solutions (1.00 µg/µL) - Stock standard solutions may be prepared from pure standard materials or purchased as certified solutions.
6.11.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in methanol and dilute to volume in a 10-mL volumetric flask. Larger volumes can be used at the convenience of the analyst. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.11.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store at 4 °C and protect from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.11.3 Stock standard solutions must be replaced after six months, or sooner if comparison with check standards indicates a problem.
6.12 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Establish chromatographic operating conditions equivalent to those given in Table 1. The HPLC system can be calibrated using the external standard technique (Section 7.2) or the internal standard technique (Section 7.3).
7.2 External standard calibration procedure:
7.2.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with mobile phase. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.2.2 Using syringe injections of 5 to 25 µL or a constant volume injection loop, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against the mass injected. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to amount injected (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.3 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask. To each calibration standard, add a known constant amount of one or more internal standards, and dilute to volume with mobile phase. One of the standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.3.2 Using syringe injections of 5 to 25 µL or a constant volume injection loop, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against concentration for each compound and internal standard. Calculate response factors (RF) for each compound using Equation 1.
RF = (As)(Cis (Ais)(Cs) Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of the parameter to be measured (µg/L).If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.
7.4 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of one or more calibration standards. If the response for any parameter varies from the predicted response by more than ±15%, a new calibration curve must be prepared for that compound. If serious loss of response occurs, polish the electrode and recalibrate.
7.5 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.9, 11.1, and 12.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Before processing any samples, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed, a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing benzidine and/or 3,3′-dichlorobenzidine at a concentration of 50 µg/mL each in methanol. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at a concentration of 50 µg/L by adding 1.00 mL of QC check sample concentrate to each of four 1-L-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter. Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at 50 µg/L or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determine background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any; or, if none (2) the larger of either 5 times higher than the expected background concentration or 50 µg/L.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100(A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 7 If spiking was performed at a concentration lower than 50 µg/L, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T)±2.44(100 S′/T)%. 7
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 1.0 mL of QC check sample concentrate (Sections 8.2.1 or 8.3.2) to 1 L of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 2. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as HPLC with a dissimilar column, gas chromatography, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 8 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C and stored in the dark from the time of collection until extraction. Both benzidine and 3,3′-dichlorobenzidine are easily oxidized. Fill the sample bottles and, if residual chlorine is present, add 80 mg of sodium thiosulfate per liter of sample and mix well. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine. 9 Field test kits are available for this purpose. After mixing, adjust the pH of the sample to a range of 2 to 7 with sulfuric acid.
9.3 If 1,2-diphenylhydrazine is likely to be present, adjust the pH of the sample to 4.0 ±0.2 to prevent rearrangement to benzidine.
9.4 All samples must be extracted within 7 days of collection. Extracts may be held up to 7 days before analysis, if stored under an inert (oxidant free) atmosphere. 2 The extract should be protected from light.
10. Sample Extraction10.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel. Check the pH of the sample with wide-range pH paper and adjust to within the range of 6.5 to 7.5 with sodium hydroxide solution or sulfuric acid.
10.2 Add 100 mL of chloroform to the sample bottle, seal, and shake 30 s to rinse the inner surface. (Caution: Handle chloroform in a well ventilated area.) Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the chloroform extract in a 250-mL separatory funnel.
10.3 Add a 50-mL volume of chloroform to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the separatory funnel. Perform a third extraction in the same manner.
10.4 Separate and discard any aqueous layer remaining in the 250-mL separatory funnel after combining the organic extracts. Add 25 mL of 1 M sulfuric acid and extract the sample by shaking the funnel for 2 min. Transfer the aqueous layer to a 250-mL beaker. Extract with two additional 25-mL portions of 1 M sulfuric acid and combine the acid extracts in the beaker.
10.5 Place a stirbar in the 250-mL beaker and stir the acid extract while carefully adding 5 mL of 0.4 M sodium tribasic phosphate. While monitoring with a pH meter, neutralize the extract to a pH between 6 and 7 by dropwise addition of 5 N sodium hydroxide solution while stirring the solution vigorously. Approximately 25 to 30 mL of 5 N sodium hydroxide solution will be required and it should be added over at least a 2-min period. Do not allow the sample pH to exceed 8.
10.6 Transfer the neutralized extract into a 250-mL separatory funnel. Add 30 mL of chloroform and shake the funnel for 2 min. Allow the phases to separate, and transfer the organic layer to a second 250-mL separatory funnel.
10.7 Extract the aqueous layer with two additional 20-mL aliquots of chloroform as before. Combine the extracts in the 250-mL separatory funnel.
10.8 Add 20 mL of reagent water to the combined organic layers and shake for 30 s.
10.9 Transfer the organic extract into a 100-mL round bottom flask. Add 20 mL of methanol and concentrate to 5 mL with a rotary evaporator at reduced pressure and 35 °C. An aspirator is recommended for use as the source of vacuum. Chill the receiver with ice. This operation requires approximately 10 min. Other concentration techniques may be used if the requirements of Section 8.2 are met.
10.10 Using a 9-in. Pasteur pipette, transfer the extract to a 15-mL, conical, screw-cap centrifuge tube. Rinse the flask, including the entire side wall, with 2-mL portions of methanol and combine with the original extract.
10.11 Carefully concentrate the extract to 0.5 mL using a gentle stream of nitrogen while heating in a 30 °C water bath. Dilute to 2 mL with methanol, reconcentrate to 1 mL, and dilute to 5 mL with acetate buffer. Mix the extract thoroughly. Cap the centrifuge tube and store refrigerated and protected from light if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial. If the sample extract requires no further cleanup, proceed with HPLC analysis (Section 12). If the sample requires further cleanup, proceed to Section 11.
10.12 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1,000-mL graduated cylinder. Record the sample volume to the nearest 5 mL.
11. Cleanup and Separation11.1 Cleanup procedures may not be necessary for a relatively clean sample matrix. If particular circumstances demand the use of a cleanup procedure, the analyst first must demonstrate that the requirements of Section 8.2 can be met using the method as revised to incorporate the cleanup procedure.
12. High Performance Liquid Chromatography12.1 Table 1 summarizes the recommended operating conditions for the HPLC. Included in this table are retention times, capacity factors, and MDL that can be achieved under these conditions. An example of the separations achieved by this HPLC column is shown in Figure 1. Other HPLC columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met. When the HPLC is idle, it is advisable to maintain a 0.1 mL/min flow through the column to prolong column life.
12.2 Calibrate the system daily as described in Section 7.
12.3 If the internal standard calibration procedure is being used, the internal standard must be added to the sample extract and mixed thoroughly immediately before injection into the instrument.
12.4 Inject 5 to 25 µL of the sample extract or standard into the HPLC. If constant volume injection loops are not used, record the volume injected to the nearest 0.05 µL, and the resulting peak size in area or peak height units.
12.5 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
12.6 If the response for a peak exceeds the working range of the system, dilute the extract with mobile phase and reanalyze.
12.7 If the measurement of the peak response for benzidine is prevented by the presence of interferences, reduce the electrode potential to + 0.6 V and reanalyze. If the benzidine peak is still obscured by interferences, further cleanup is required.
13. Calculations13.1 Determine the concentration of individual compounds in the sample.
13.1.1 If the external standard calibration procedure is used, calculate the amount of material injected from the peak response using the calibration curve or calibration factor determined in Section 7.2.2. The concentration in the sample can be calculated from Equation 2.
Equation 2 where: A = Amount of material injected (ng). Vi = Volume of extract injected (µL). Vt = Volume of total extract (µL). Vs = Volume of water extracted (mL).13.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.3.2 and Equation 3.
Equation 3 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).13.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
14. Method Performance14.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Table 1 were obtained using reagent water. 10 Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
14.2 This method has been tested for linearity of spike recovery from reagent water and has been demonstrated to be applicable over the concentration range from 7 × MDL to 3000 × MDL. 10
14.3 This method was tested by 17 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 1.0 to 70 µg/L. 11 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. 40 CFR part 136, appendix B.
2. “Determination of Benzidines in Industrial and Muncipal Wastewaters,” EPA 600/4-82-022, National Technical Information Service, PB82-196320, Springfield, Virginia 22161, April 1982.
3. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia.
4. “Carcinogens - Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
5. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
7. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
8. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
9. “Methods 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric, DPD) for Chlorine Total Residual,” Methods for Chemical Analysis of Water and Wastes, EPA-600/4-79-020, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1979.
10. “EPA Method Study 15, Method 605 (Benzidines),” EPA 600/4-84-062, National Technical Information Service, PB84-211176, Springfield, Virginia 22161, June 1984.
11. “EPA Method Validation Study 15, Method 605 (Benzidines),” Report for EPA Contract 68-03-2624 (In preparation).
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Column capacity factor (k′) | Method detection limit (µg/L) |
|---|---|---|---|
| Benzidine | 6.1 | 1.44 | 0.08 |
| 3,3′-Dichlorobenzidine | 12.1 | 3.84 | 0.13 |
HPLC Column conditions: Lichrosorb RP-2, 5 micron particle size, in a 25 cm × 4.6 mm ID stainless steel column. Mobile Phase: 0.8 mL/min of 50% acetonitrile/50% 0.1M pH 4.7 acetate buffer. The MDL were determined using an electrochemical detector operated at + 0.8 V.
Table 2 - QC Acceptance Criteria - Method 605
| Parameter | Test conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps (percent) |
|---|---|---|---|---|
| Benzidine | 50 | 18.7 | 9.1-61.0 | D-140 |
| 3.3′-Dichlorobenzidine | 50 | 23.6 | 18.7-50.0 | 5-128 |
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
D = Detected; result must be greater than zero.
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 605
| Parameter | Accuracy, as recovery, X′(µg/L) | Single analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| Benzidine | 0.70C + 0.06 | 0.28X + 0.19 | 0.40X + 0.18 |
| 3,3′-Dichlorobenzidine | 0.66C + 0.23 | 0.39X −0.05 | 0.38X + 0.02 |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
 Method
606 - Phthalate Ester 1. Scope and Application
Method
606 - Phthalate Ester 1. Scope and Application
1.1 This method covers the determination of certain phthalate esters. The following parameters can be determined by this method:
| Parameter | STORET No. | CAS No. |
|---|---|---|
| Bis(2-ethylhexyl) phthalate | 39100 | 117-81-7 |
| Butyl benzyl phthalate | 34292 | 85-68-7 |
| Di-n-butyl phthalate | 39110 | 84-74-2 |
| Diethyl phthalate | 34336 | 84-66-2 |
| Dimethyl phthalate | 34341 | 131-11-3 |
| Di-n-octyl phthalate | 34596 | 117-84-0 |
1.2 This is a gas chromatographic (GC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compounds above, compound identifications should be supported by at least one additional qualitative technique. This method describes analytical conditions for a second gas chromatographic column that can be used to confirm measurements made with the primary column. Method 625 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for all of the parameters listed above, using the extract produced by this method.
1.3 The method detection limit (MDL, defined in Section 14.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.4 The sample extraction and concentration steps in this method are essentially the same as in Methods 608, 609, 611, and 612. Thus, a single sample may be extracted to measure the parameters included in the scope of each of these methods. When cleanup is required, the concentration levels must be high enough to permit selecting aliquots, as necessary, to apply appropriate cleanup procedures. The analyst is allowed the latitude, under Section 12, to select chromatographic conditions appropriate for the simultaneous measurement of combinations of these parameters.
1.5 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.6 This method is restricted to use by or under the supervision of analysts experienced in the use of a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is extracted with methylene chloride using a separatory funnel. The methylene chloride extract is dried and exchanged to hexane during concentration to a volume of 10 mL or less. The extract is separated by gas chromatography and the phthalate esters are then measured with an electron capture detector. 2
2.2 Analysis for phthalates is especially complicated by their ubiquitous occurrence in the environment. The method provides Florisil and alumina column cleanup procedures to aid in the elimination of interferences that may be encountered.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardware that lead to discrete artifacts and/or elevated baselines in gas chromatograms. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 3 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Some thermally stable materials, such as PCBs, may not be eliminated by this treatment. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Thorough rinsing with such solvents usually eliminates PCB interference. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to minimize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Phthalate esters are contaminants in many products commonly found in the laboratory. It is particularly important to avoid the use of plastics because phthalates are commonly used as plasticizers and are easily extracted from plastic materials. Serious phthalate contamination can result at any time, if consistent quality control is not practiced. Great care must be experienced to prevent such contamination. Exhaustive cleanup of reagents and glassware may be required to eliminate background phthalate contamination. 4 5
3.3 Matrix interferences may be caused by contaminants that are co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. The cleanup procedures in Section 11 can be used to overcome many of these interferences, but unique samples may require additional cleanup approaches to achieve the MDL listed in Table 1.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 6 8 for the information of the analyst.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1-L or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flow meter is required to collect flow proportional composites.
5.2 Glassware (All specifications are suggested. Catalog numbers are included for illustration only).
5.2.1 Separatory funnel - 2-L, with Teflon stopcock.
5.2.2 Drying column - Chromatographic column, approximately 400 mm long × 19 mm ID, with coarse frit filter disc.
5.2.3 Chromatographic column - 300 mm long × 10 mm ID, with Teflon stopcock and coarse frit filter disc at bottom (Kontes K-420540-0213 or equivalent).
5.2.4 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes K-570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. Ground glass stopper is used to prevent evaporation of extracts.
5.2.5 Evaporative flask, Kuderna-Danish - 500-mL (Kontes K-570001-0500 or equivalent). Attach to concentrator tube with springs.
5.2.6 Snyder column, Kuderna-Danish - Three-ball macro (Kontes K-503000-0121 or equivalent).
5.2.7 Snyder column, Kuderna-Danish - Two-ball micro (Kontes K-569001-0219 or equivalent).
5.2.8 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.3 Boiling chips - Approximately 10/40 mesh. Heat to 400 °C for 30 min or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 Balance - Analytical, capable of accurately weighing 0.0001 g.
5.6 Gas chromatograph - An analytical system complete with gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.6.1 Column 1 - 1.8 m long × 4 mm ID glass, packed with 1.5% SP-2250/1.95% SP-2401 Supelcoport (100/120 mesh) or equivalent. This column was used to develop the method performance statemelts in Section 14. Guidelines for the use of alternate column packings are provided in Section 12.1.
5.6.2 Column 2 - 1.8 m long × 4 mm ID glass, packed with 3% OV-1 on Supelcoport (100/120 mesh) or equivalent.
5.6.3 Detector - Electron capture detector. This detector has proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1), and was used to develop the method performance statements in Section 14. Guidelines for the use of alternate detectors are provided in Section 12.1.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.2 Acetone, hexane, isooctane, methylene chloride, methanol - Pesticide quality or equivalent.
6.3 Ethyl ether - nanograde, redistilled in glass if necessary.
6.3.1 Ethyl ether must be shown to be free of peroxides before it is used as indicated by EM Laboratories Quant test strips. (Available from Scientific Products Co., Cat. No. P1126-8, and other suppliers.)
6.3.2 Procedures recommended for removal of peroxides are provided with the test strips. After cleanup, 20 mL of ethyl alcohol preservative must be added to each liter of ether.
6.4 Sodium sulfate - (ACS) Granular, anhydrous. Several levels of purification may be required in order to reduce background phthalate levels to an acceptable level: 1) Heat 4 h at 400 °C in a shallow tray, 2) Heat 16 h at 450 to 500 °C in a shallow tray, 3) Soxhlet extract with methylene chloride for 48 h.
6.5 Florisil - PR grade (60/100 mesh). Purchase activated at 1250 °F and store in the dark in glass containers with ground glass stoppers or foil-lined screw caps. To prepare for use, place 100 g of Florisil into a 500-mL beaker and heat for approximately 16 h at 40 °C. After heating transfer to a 500-mL reagent bottle. Tightly seal and cool to room temperature. When cool add 3 mL of reagent water. Mix thoroughly by shaking or rolling for 10 min and let it stand for at least 2 h. Keep the bottle sealed tightly.
6.6 Alumina - Neutral activity Super I, W200 series (ICN Life Sciences Group, No. 404583). To prepare for use, place 100 g of alumina into a 500-mL beaker and heat for approximately 16 h at 400 °C. After heating transfer to a 500-mL reagent bottle. Tightly seal and cool to room temperature. When cool add 3 mL of reagent water. Mix thoroughly by shaking or rolling for 10 min and let it stand for at least 2 h. Keep the bottle sealed tightly.
6.7 Stock standard solutions (1.00 µg/µL) - Stock standard solutions can be prepared from pure standard materials or purchased as certified solutions.
6.7.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in isooctane and dilute to volume in a 10-mL volumetric flask. Larger volumes can be used at the convenience of the analyst. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.7.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store at 4 °C and protect from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.7.3 Stock standard solutions must be replaced after six months, or sooner if comparison with check standards indicates a problem.
6.8 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Establish gas chromatograph operating conditions equivalent to those given in Table 1. The gas chromatographic system can be calibrated using the external standard technique (Section 7.2) or the internal standard technique (Section 7.3).
7.2 External standard calibration procedure:
7.2.1 Prepared calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with isooctane. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.2.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against the mass injected. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to amount injected (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.3 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flash. To each calibration standard, add a known constant amount of one or more internal standards, and dilute to volume with isooctane. One of the standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.3.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against concentration for each compound and internal standard. Calculate response factors (RF) for each compound using Equation 1.
RF = (As)(Cis (Ais)(Cs) Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of the parameter to be measured (µg/L).If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.
7.4 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of one or more calibration standards. If the response for any parameter varies from the predicted response by more than ±15%, a new calibration curve must be prepared for that compound.
7.5 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.4, 11.1, and 12.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Before processing any samples, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed, a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality contrml (QC) check sample concentrate is required containing each parameter of interest at the following concentrations in acetone: butyl benzyl phthalate, 10 µg/mL; bis(2-ethylhexyl) phthalate, 50 µg/mL; di-n-octyl phthalate, 50 µg/mL; any other phthlate, 25 µg/mL. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agancy, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at the test concentrations shown in Table 2 by adding 1.00 mL of QC check sample concentrate to each of four 1-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter. Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at the test concentration in Section 8.2.2 or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determine background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any; or, if none (2) the larger of either 5 times higher than the expected background concentration or the test concentration in Section 8.2.2.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100(A-B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 9 If spiking was performed at a concentration lower than the test concentration in Section 8.2.2, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T)±2.44(100 S′/T)%. 9
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 1.0 mL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 1 L of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 2. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 10 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C from the time of collection until extraction.
9.3 All samples must be extracted within 7 days of collection and completely analyzed within 40 days of extraction. 2
10. Sample Extraction10.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel.
10.2 Add 60 mL of methylene chloride to the sample bottle, seal, and shake 30 s to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min. with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phrase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the methylene chloride extract in a 250-mL Erlenmeyer flask.
10.3 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.4 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentrator devices or techniques may be used in place of the K-D concentrator if the requirements of Section 8.2 are met.
10.5 Pour the combined extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the Erlenmeyer flask and column with 20 to 30 mL of methylene chloride to complete the quantitative transfer.
10.6 Add one or two clean boiling chips to the evaporative flask and attach a three-ball Snyder column. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
10.7 Increase the temperature of the hot water bath to about 80 °C. Momentarily remove the Snyder column, add 50 mL of hexane and a new boiling chip, and reattach the Snyder column. Concentrate the extract as in Section 10.6, except use hexane to prewet the column. The elapsed time of concentration should be 5 to 10 min.
10.8 Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of hexane. A 5-mL syringe is recommended for this operation. Adjust the extract volume to 10 mL. Stopper the concentrator tube and store refrigerated if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial. If the sample extract requires no further cleanup, proceed with gas chromatographic analysis (Section 12). If the sample requires further cleanup, proceed to Section 11.
10.9 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1000-mL graduated cylinder. Record the sample volume to the nearest 5 mL.
11. Cleanup and Separation11. Cleanup procedures may not be necessary for a relatively clean sample matrix. If particular circumstances demand the use of a cleanup procedure, the analyst may use either procedure below or any other appropriate procedure. However, the analyst first must demonstrate that the requirements of Section 8.2 can be met using the method as revised to incorporate the cleanup procedure.
11.2 If the entire extract is to be cleaned up by one of the following procedures, it must be concentrated to 2.0 mL. To the concentrator tube in Section 10.8, add a clean boiling chip and attach a two-ball micro-Snyder column. Prewet the column by adding about 0.5 mL of hexane to the top. Place the micro-K-D apparatus on a hot water bath (80 °C) so that the concentrator tube is partially immersed in the hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 5 to 10 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood. When the apparent volume of liquid reaches about 0.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min. Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with 0.2 mL of hexane. Adjust the final volume to 2.0 mL and proceed with one of the following cleanup procedures.
11.3 Florisil column cleanup for phthalate esters:
11.3.1 Place 10 g of Florisil into a chromatographic column. Tap the column to settle the Florisil and add 1 cm of anhydrous sodium sulfate to the top.
11.3.2 Preelute the column with 40 mL of hexane. The rate for all elutions should be about 2 mL/min. Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the 2-mL sample extract onto the column using an additional 2 mL of hexane to complete the transfer. Just prior to exposure of the sodium sulfate layer to the air, add 40 mL of hexane and continue the elution of the column. Discard this hexane eluate.
11.3.3 Next, elute the column with 100 mL of 20% ethyl ether in hexane (V/V) into a 500-mL K-D flask equipped with a 10-mL concentrator tube. Concentrate the collected fraction as in Section 10.6. No solvent exchange is necessary. Adjust the volume of the cleaned up extract to 10 mL in the concentrator tube and analyze by gas chromatography (Section 12).
11.4 Alumina column cleanup for phthalate esters:
11.4.1 Place 10 g of alumina into a chromatographic column. Tap the column to settle the alumina and add 1 cm of anhydrous sodium sulfate to the top.
11.4.2 Preelute the column with 40 mL of hexane. The rate for all elutions should be about 2 mL/min. Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the 2-mL sample extract onto the column using an additional 2 mL of hexane to complete the transfer. Just prior to exposure of the sodium sulfate layer to the air, add 35 mL of hexane and continue the elution of the column. Discard this hexane eluate.
11.4.3 Next, elute the column with 140 mL of 20% ethyl ether in hexane (V/V) into a 500-mL K-D flask equipped with a 10-mL concentrator type. Concentrate the collected fraction as in Section 10.6. No solvent exchange is necessary. Adjust the volume of the cleaned up extract to 10 mL in the concentrator tube and analyze by gas chromatography (Section 12).
12. Gas Chromatography12.1 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and MDL that can be achieved under these conditions. Examples of the separations achieved by Column 1 are shown in Figures 1 and 2. Other packed or capillary (open-tubular) columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
12.2 Calibrate the system daily as described in Section 7.
12.3 If the internal standard calibration procedure is being used, the internal staldard must be added to the sample extract and mixed thoroughly immediately before injection into the gas chromatograph.
12.4 Inject 2 to 5 µL of the sample extract or standard into the gas-chromatograph using the solvent-flush technique. 11 Smaller (1.0 µL) volumes may be injected if automatic devices are employed. Record the volume injected to the nearest 0.05 µL, and the resulting peak size in area or peak height units.
12.5 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
12.6 If the response for a peak exceeds the working range of the system, dilute the extract and reanalyze.
12.7 If the measurement of the peak response is prevented by the presence of interferences, further cleanup is required.
13. Calculations13.1 Determine the concentration of individual compounds in the sample.
13.1.1 If the external standard calibration procedure is used, calculate the amount of material injected from the peak response using the calibration curve or calibration factor determined in Section 7.2.2. The concentration in the sample can be calculated from Equation 2.
Equation 2 where: A = Amount of material injected (ng). Vi = Volume of extract injected (µL). Vt = Volume of total extract (µL). Vs = Volume of water extracted (mL).13.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.3.2 and Equation 3.
Equation 3 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).13.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
14. Method Performance14.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Table 1 were obtained using reagent water. 12 Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
14.2 This method has been tested for linearity of spike recovery from reagent water and has been demonstrated to be applicable over the concentration range from 5 × MDL to 1000 × MDL with the following exceptions: dimethyl and diethyl phthalate recoveries at 1000 × MDL were low (70%); bis-2-ethylhexyl and di-n-octyl phthalate recoveries at 5 × MDL were low (60%). 12
14.3 This method was tested by 16 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 0.7 to 106 µg/L. 13 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. 40 CFR part 136, appendix B.
2. “Determination of Phthalates in Industrial and Muncipal Wastewaters,” EPA 600/4-81-063, National Technical Information Service, PB81-232167, Springfield, Virginia 22161, July 1981.
3. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia.
4. Giam, C.S., Chan, H.S., and Nef, G.S. “Sensitive Method for Determination of Phthalate Ester Plasticizers in Open-Ocean Biota Samples,” Analytical Chemistry, 47, 2225 (1975).
5. Giam, C.S., and Chan, H.S. “Control of Blanks in the Analysis of Phthalates in Air and Ocean Biota Samples,” U.S. National Bureau of Standards, Special Publication 442, pp. 701-708, 1976.
6. “Carcinogens - Working with Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
7. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
8. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
9. Provost L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
10. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
11. Burke, J.A. “Gas Chromatography for Pesticide Residue Analysis; Some Practical Aspects,” Journal of the Association of Official Analytical Chemists, 48, 1037 (1965).
12. “Method Detection Limit and Analytical Curve Studies, EPA Methods 606, 607, and 608,” Special letter report for EPA Contract 68-03-2606, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, June 1980.
13. “EPA Method Study 16 Method 606 (Phthalate Esters),” EPA 600/4-84-056, National Technical Information Service, PB84-211275, Springfield, Virginia 22161, June 1984.
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Method detection limit (µg/L) | |
|---|---|---|---|
| Column 1 | Column 2 | ||
| Dimethyl phthalate | 2.03 | 0.95 | 0.29 |
| Diethyl phthalate | 2.82 | 1.27 | 0.49 |
| Di-n-butyl phthalate | 8.65 | 3.50 | 0.36 |
| Butyl benzyl phthalate | a 6.94 | a 5.11 | 0.34 |
| Bis(2-ethylhexyl) phthalate | a 8.92 | a 10.5 | 2.0 |
| Di-n-octyl phthalate | a 16.2 | a 18.0 | 3.0 |
Column 1 conditions: Supelcoport (100/120 mesh) coated with 1.5% SP-2250/1.95% SP-2401 packed in a 1.8 m long × 4 mm ID glass column with 5% methane/95% argon carrier gas at 60 mL/min flow rate. Column temperature held isothermal at 180 °C, except where otherwise indicated.
Column 2 conditions: Supelcoport (100/120 mesh) coated with 3% OV-1 packed in a 1.8 m long × 4 mm ID glass column with 5% methane/95% argon carrier gas at 60 mL/min flow rate. Column temperature held isothermal at 200 °C, except where otherwise indicated.
a 220 °C column temperature.
Table 2 - QC Acceptance Criteria - Method 606
| Parameter | Test conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps (percent) |
|---|---|---|---|---|
| Bis(2-ethylhexyl) phthalate | 50 | 38.4 | 1.2-55.9 | D-158 |
| Butyl benzyl phthalate | 10 | 4.2 | 5.7-11.0 | 30-136 |
| Di-n-butyl phthalate | 25 | 8.9 | 10.3-29.6 | 23-136 |
| Diethyl phthalate | 25 | 9.0 | 1.9-33.4 | D-149 |
| Dimethyl phathalate | 25 | 9.5 | 1.3-35.5 | D-156 |
| Di-n-octyl phthalate | 50 | 13.4 | D-50.0 | D-114 |
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
D = Detected; result must be greater than zero.
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 606
| Parameter | Accuracy, as recovery, X′ (µg/L) | Single analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| Bis(2-ethylhexyl) phthalate | 0.53C + 2.02 | 0.80X −2.54 | 0.73X −0.17 |
| Butyl benzyl phthalate | 0.82C + 0.13 | 0.26X + 0.04 | 0.25X + 0.07 |
| Di-n-butyl phthalate | 0.79C + 0.17 | 0.23X + 0.20 | 0.29X + 0.06 |
| Diethyl phthalate | 0.70C + 0.13 | 0.27X + 0.05 | 0.45X + 0.11 |
| Dimethyl phthalate | 0.73C + 0.17 | 0.26X + 0.14 | 0.44X + 0.31 |
| Di-n-octyl phthalate | 0.35C−0.71 | 0.38X + 0.71 | 0.62X + 0.34 |
X ′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
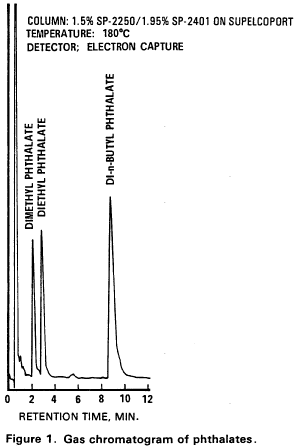
 Method 607 -
Nitrosamines 1. Scope and Application
Method 607 -
Nitrosamines 1. Scope and Application
1.1 This method covers the determination of certain nitrosamines. The following parameters can be determined by this method:
| Parameter | Storet No. | CAS No. |
|---|---|---|
| N-Nitrosodimethylamine | 34438 | 62-75-9 |
| N-Nitrosodiphenylamine | 34433 | 86-30-6 |
| N-Nitrosodi-n-propylamine | 34428 | 621-64-7 |
1.2 This is a gas chromatographic (GC) method applicable to the determination of the parameters listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compmunds above, compound identifications should be supported by at least one additional qualitative technique. This method describes analytical conditimns for a second gas chromatographic column that can be used to confirm measurements made with the primary column. Method 625 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for N-nitrosodi-n-propylamine. In order to confirm the presence of N-nitrosodiphenylamine, the cleanup procedure specified in Section 11.3 or 11.4 must be used. In order to confirm the presence of N-nitrosodimethylamine by GC/MS, Column 1 of this method must be substituted for the column recommended in Method 625. Confirmation of these parameters using GC-high resolution mass spectrometry or a Thermal Energy Analyzer is also recommended. 1 2
1.3 The method detection limit (MDL, defined in Section 14.1) 3 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.4 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.5 This method is restricted to use by or under the supervision of analysts experienced in the use of a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is extracted with methylene chloride using a separatory funnel. The methylene chloride extract is washed with dilute hydrochloric acid to remove free amines, dried, and concentrated to a volume of 10 mL or less. After the extract has been exchanged to methanol, it is separated by gas chromatography and the parameters are then measured with a nitrogen-phosphorus detector. 4
2.2 The method provides Florisil and alumina column cleanup procedures to separate diphenylamine from the nitrosamines and to aid in the elimination of interferences that may be encountered.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardware that lead to discrete artifacts and/or elevated baselines in gas chromatograms. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 5 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to minimize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Matrix interferences may be caused by contaminants that are co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. The cleanup procedures in Section 11 can be used to overcome many of these interferences, but unique samples may require additional cleanup approaches to achieve the MDL listed in Table 1.
3.3 N-Nitrosodiphenylamine is reported 6-9 to undergo transnitrosation reactions. Care must be exercised in the heating or concentrating of solutions containing this compound in the presence of reactive amines.
3.4 The sensitive and selective Thermal Energy Analyzer and the reductive Hall detector may be used in place of the nitrogen-phosphorus detector when interferences are encountered. The Thermal Energy Analyzer offers the highest selectivity of the non-MS detectors.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 10-12 for the information of the analyst.
4.2 These nitrosamines are known carcinogens, 13-17 therefore, utmost care must be exercised in the handling of these materials. Nitrosamine reference standards and standard solutions should be handled and prepared in a ventilated glove box within a properly ventilated room.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1-L or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flowmeter is required to collect flow proportional composites.
5.2 Glassware (All specifications are suggested. Catalog numbers are included for illustration only.):
5.2.1 Separatory funnels - 2-L and 250-mL, with Teflon stopcock.
5.2.2 Drying column - Chromatographic column, approximately 400 mm long × 19 mm ID, with coarse frit filter disc.
5.2.3 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes K-570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. Ground glass stopper is used to prevent evaporation of extracts.
5.2.4 Evaporative flask, Kuderna-Danish - 500-mL (Kontes K-570001-0500 or equivalent). Attach to concentrator tube with springs.
5.2.5 Snyder column, Kuderna-Danish - Three-ball macro (Kontes K-503000-0121 or equivalent).
5.2.6 Snyder column, Kuderna-Danish - Two-ball micro (Kontes K-569001-0219 or equivalent).
5.2.7 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.2.8 Chromatographic column - Approximately 400 mm long × 22 mm ID, with Teflon stopcock and coarse frit filter disc at bottom (Kontes K-420540-0234 or equivalent), for use in Florisil column cleanup procedure.
5.2.9 Chromatographic column - Approximately 300 mm long × 10 mm ID, with Teflon stopcock and coarse frit filter disc at bottom (Kontes K-420540-0213 or equivalent), for use in alumina column cleanup procedure.
5.3 Boiling chips - Approximately 10/40 mesh. Heat to 400 °C for 30 min or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 Balance - Analytical, capable of accurately weighing 0.0001 g.
5.6 Gas chromatograph - An analytical system complete with gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.6.1 Column 1 - 1.8 m long × 4 mm ID glass, packed with 10% Carbowax 20 M/2% KOH on Chromosorb W-AW (80/100 mesh) or equivalent. This column was used to develop the method performance statements in Section 14. Guidelines for the use of alternate column packings are provided in Section 12.2.
5.6.2 Column 2 - 1.8 m long × 4 mm ID glass, packed with 10% SP-2250 on Supel-coport (100/120 mesh) or equivalent.
5.6.3 Detector - Nitrogen-phosphorus, reductive Hall, or Thermal Energy Analyzer detector. 1 2 These detectors have proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1). A nitrogen-phosphorus detector was used to develop the method performance statements in Section 14. Guidelines for the use of alternate detectors are provided in Section 12.2.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.2 Sodium hydroxide solution (10 N) - Dissolve 40 g of NaOH (ACS) in reagent water and dilute to 100 ml.
6.3 Sodium thiosulfate - (ACS) Granular.
6.4 Sulfuric acid (1 + 1) - Slowly, add 50 mL of H2SO4 (ACS, sp. gr. 1.84) to 50 mL of reagent water.
6.5 Sodium sulfate - (ACS) Granular, anhydrous. Purify by heating at 400 °C for 4 h in a shallow tray.
6.6 Hydrochloric acid (1 + 9) - Add one volume of concentrated HCl (ACS) to nine volumes of reagent water.
6.7 Acetone, methanol, methylene chloride, pentane - Pesticide quality or equivalent.
6.8 Ethyl ether - Nanograde, redistilled in glass if necessary.
6.8.1 Ethyl ether must be shown to be free of peroxides before it is used as indicated by EM Laboratories Quant test strips. (Available from Scientific Products Co., Cat No. P1126-8, and other suppliers.)
6.8.2 Procedures recommended for removal of peroxides are provided with the test strips. After cleanup, 20 mL of ethyl alcohol preservative must be added to each liter of ether.
6.9 Florisil - PR grade (60/100 mesh). Purchase activated at 1250 °F and store in the dark in glass containers with ground glass stoppers or foil-lined screw caps. Before use, activate each batch at least 16 h at 130 °C in a foil-covered glass container and allow to cool.
6.10 Alumina - Basic activity Super I, W200 series (ICN Life Sciences Group, No. 404571, or equivalent). To prepare for use, place 100 g of alumina into a 500-mL reagent bottle and add 2 mL of reagent water. Mix the alumina preparation thoroughly by shaking or rolling for 10 min and let it stand for at least 2 h. The preparation should be homogeneous before use. Keep the bottle sealed tightly to ensure proper activity.
6.11 Stock standard solutions (1.00 µg/µL) - Stock standard solutions can be prepared from pure standard materials or purchased as certified solutions.
6.11.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in methanol and dilute to volume in a 10-mL volumetric flask. Larger volumes can be used at the convenience of the analyst. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.11.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store at 4 °C and protect from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.11.3 Stock standard solutions must be replaced after six months, or sooner if comparison with check standards indicates a problem.
6.12 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Establish gas chromatographic operating conditions equivalent to those given in Table 1. The gas chromatographic system can be calibrated using the external standard technique (Section 7.2) or the internal standard technique (Section 7.3).
7.2 External standard calibration procedure:
7.2.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with methanol. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.2.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against the mass injected. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to amount injected (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.3 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask. To each calibration standard, add a known constant amount of one or more internal standards, and dilute to volume with methanol. One of the standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.3.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against concentration for each compound and internal standard. Calculate response factors (RF) for each compound using Equation 1.
RF = (As)(Cis (Ais)(Cs) Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of the parameter to be measured (µg/L).If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.
7.4 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of one or more calibration standards. If the response for any parameter varies from the predicted response by more than ±15%, a new calibration curve must be prepared for that compound.
7.5 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.4, 11.1, and 12.2) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Before processing any samples, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed, a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest at a concentration of 20 µg/mL in methanol. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at a concentration of 20 µg/L by adding 1.00 mL of QC check sample concentrate to each of four 1-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter. Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at 20 µg/L or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determine background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any; or, if none (2) the larger of either 5 times higher than the expected background concentration or 20 µg/L.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100(A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were caluclated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 18 If spiking was performed at a concentration lower than 20 µg/L, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria caluclated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T) ±2.44(100 S′/T)%. 18
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 1.0 mL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 1 L of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 2. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 19 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C from the time of collection until extraction. Fill the sample bottles and, if residual chlorine is present, add 80 mg of sodium thiosulfate per liter of sample and mix well. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine. 20 Field test kits are available for this purpose. If N-nitrosodiphenylamine is to be determined, adjust the sample pH to 7 to 10 with sodium hydroxide solution or sulfuric acid.
9.3 All samples must be extracted within 7 days of collection and completely analyzed within 40 days of extraction. 4
9.4 Nitrosamines are known to be light sensitive. 7 Samples should be stored in amber or foil-wrapped bottles in order to minimize photolytic decomposition.
10. Sample Extraction10.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel. Check the pH of the sample with wide-range pH paper and adjust to within the range of 5 to 9 with sodium hydroxide solution or sulfuric acid.
10.2 Add 60 mL of methylene chloride to the sample bottle, seal, and shake 30 s to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the methylene chloride extract in a 250-mL Erlenmeyer flask.
10.3 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.4 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator if the requirements of Section 8.2 are met.
10.5 Add 10 mL of hydrochloric acid to the combined extracts and shake for 2 min. Allow the layers to separate. Pour the combined extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the Erlenmeyer flask and column with 20 to 30 mL of methylene chloride to complete the quantitative transfer.
10.6 Add one or two clean boiling chips to the evaporative flask and attach a three-ball Snyder column. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
10.7 Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of methylene chloride. A 5-mL syringe is recommended for this operation. Stopper the concentrator tube and store refrigerated if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial. If N-nitrosodiphenylamine is to be measured by gas chromatography, the analyst must first use a cleanup column to eliminate diphenylamine interference (Section 11). If N-nitrosodiphenylamine is of no interest, the analyst may proceed directly with gas chromatographic analysis (Section 12).
10.8 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1000-
mL graduated cylinder. Record the sample volume to the nearest 5 mL. 11. Cleanup and Separation11.1 Cleanup procedures may not be necessary for a relatively clean sample matrix. If particular circumstances demand the use of a cleanup procedure, the analyst may use either procedure below or any other appropriate procedure. However, the analyst first must demonstrate that the requirements of Section 8.2 can be met using the method as revised to incorporate the cleanup procedure. Diphenylamine, if present in the original sample extract, must be separated from the nitrosamines if N-nitrosodiphenylamine is to be determined by this method.
11.2 If the entire extract is to be cleaned up by one of the following procedures, it must be concentrated to 2.0 mL. To the concentrator tube in Section 10.7, add a clean boiling chip and attach a two-ball micro-Snyder column. Prewet the column by adding about 0.5 mL of methylene chloride to the top. Place the micr-K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 5 to 10 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood. When the apparent volume of liquid reaches about 0.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min. Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with 0.2 mL of methylene chloride. Adjust the final volume to 2.0 mL and proceed with one of the following cleanup procedures.
11.3 Florisil column cleanup for nitrosamines:
11.3.1 Place 22 g of activated Florisil into a 22-mm ID chromatographic column. Tap the column to settle the Florisil and add about 5 mm of anhydrous sodium sulfate to the top.
11.3.2 Preelute the column with 40 mL of ethyl ether/pentane (15 + 85)(V/V). Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the 2-mL sample extract onto the column using an additional 2 mL of pentane to complete the transfer.
11.3.3 Elute the column with 90 mL of ethyl ether/pentane (15 + 85)(V/V) and discard the eluate. This fraction will contain the diphenylamine, if it is present in the extract.
11.3.4 Next, elute the column with 100 mL of acetone/ethyl ether (5 + 95)(V/V) into a 500-mL K-D flask equipped with a 10-mL concentrator tube. This fraction will contain all of the nitrosamines listed in the scope of the method.
11.3.5 Add 15 mL of methanol to the collected fraction and concentrate as in Section 10.6, except use pentane to prewet the column and set the water bath at 70 to 75 °C. When the apparatus is cool, remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of pentane. Analyze by gas chromatography (Section 12).
11.4 Alumina column cleanup for nitrosamines:
11.4.1 Place 12 g of the alumina preparation (Section 6.10) into a 10-mm ID chromatographic column. Tap the column to settle the alumina and add 1 to 2 cm of anhydrous sodium sulfate to the top.
11.4.2 Preelute the column with 10 mL of ethyl ether/pentane (3 + 7)(V/V). Discard the eluate (about 2 mL) and just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the 2 mL sample extract onto the column using an additional 2 mL of pentane to complete the transfer.
11.4.3 Just prior to exposure of the sodium sulfate layer to the air, add 70 mL of ethyl ether/pentane (3 + 7)(V/V). Discard the first 10 mL of eluate. Collect the remainder of the eluate in a 500-mL K-D flask equipped with a 10 mL concentrator tube. This fraction contains N-nitrosodiphenylamine and probably a small amount of N-nitrosodi-n-propylamine.
11.4.4 Next, elute the column with 60 mL of ethyl ether/pentane (1 + 1)(V/V), collecting the eluate in a second K-D flask equipped with a 10-mL concentrator tube. Add 15 mL of methanol to the K-D flask. This fraction will contain N-nitrosodimethylamine, most of the N-nitrosodi-n-propylamine and any diphenylamine that is present.
11.4.5 Concentrate both fractions as in Section 10.6, except use pentane to prewet the column. When the apparatus is cool, remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of pentane. Analyze the fractions by gas chromatography (Section 12).
12. Gas Chromatography12.1 N-nitrosodiphenylamine completely reacts to form diphenylamine at the normal operating temperatures of a GC injection port (200 to 250 °C). Thus, N-nitrosodiphenylamine is chromatographed and detected as diphenylamine. Accurate determination depends on removal of diphenylamine that may be present in the original extract prior to GC analysis (See Section 11).
12.2 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and MDL that can be achieved under these conditions. Examples of the separations achieved by Column 1 are shown in Figures 1 and 2. Other packed or capillary (open-tubular) columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
12.3 Calibrate the system daily as described in Section 7.
12.4 If the extract has not been subjected to one of the cleanup procedures in Section 11, it is necessary to exchange the solvent from methylene chloride to methanol before the thermionic detector can be used. To a 1 to 10-mL volume of methylene chloride extract in a concentrator tube, add 2 mL of methanol and a clean boiling chip. Attach a two-ball micro-Snyder column to the concentrator tube. Prewet the column by adding about 0.5 mL of methylene chloride to the top. Place the micro-K-D apparatus on a boiling (100 °C) water bath so that the concentrator tube is partially immersed in the hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 5 to 10 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood. When the apparent volume of liquid reaches about 0.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min. Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with 0.2 mL of methanol. Adjust the final volume to 2.0 mL.
12.5 If the internal standard calibration procedure is being used, the internal standard must be added to the sample extract and mixed thoroughly immediately before injection into the gas chromatograph.
12.6 Inject 2 to 5 µL of the sample extract or standard into the gas chromatograph using the solvent-flush technique. 21 Smaller (1.0 µL) volumes may be injected if automatic devices are employed. Record the volume injected to the nearest 0.05 µL, and the resulting peak size in area or peak height units.
12.7 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
12.8 If the response for a peak exceeds the working range of the system, dilute the extract and reanalyze.
12.9 If the measurement of the peak response is prevented by the presence of interferences, further cleanup is required.
13. Calculations13.1 Determine the concentration of individual compounds in the sample.
13.1.1 If the external standard calibration procedure is used, calculate the amount of material injected from the peak response using the calibration curve or calibration factor determined in Section 7.2.2. The concentration in the sample can be calculated from Equation 2.
Equation 2 where: A = Amount of material injected (ng). Vi = Volume of extract injected (µL). Vt = Volume of total extract (µL). Vs = Volume of water extracted (mL).13.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.3.2 and Equation 3.
Equation 3 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).13.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
14. Method Performance14.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 3 The MDL concentrations listed in Table 1 were obtained using reagent water. 22 Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
14.2 This method has been tested for linearity of spike recovery from reagent water and has been demonstrated to be applicable over the concentration range from 4 × MDL to 1000 × MDL. 22
14.3 This method was tested by 17 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 0.8 to 55 µg/L. 23 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. Fine, D.H., Lieb, D., and Rufeh, R. “Principle of Operation of the Thermal Energy Analyzer for the Trace Analysis of Volatile and Non-volatile N-nitroso Compounds,” Journal of Chromatography, 107, 351 (1975).
2. Fine, D.H., Hoffman, F., Rounbehler, D.P., and Belcher, N.M. “Analysis of N-nitroso Compounds by Combined High Performance Liquid Chromatography and Thermal Energy Analysis,” Walker, E.A., Bogovski, P. and Griciute, L., Editors, N-nitroso Compounds - Analysis and Formation, Lyon, International Agency for Research on Cancer (IARC Scientific Publications No. 14), pp. 43-50 (1976).
3. 40 CFR part 136, appendix B.
4. “Determination of Nitrosamines in Industrial and Municipal Wastewaters,” EPA 600/4-82-016, National Technical Information Service, PB82-199621, Springfield, Virginia 22161, April 1982.
5. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia.
6. Buglass, A.J., Challis, B.C., and Osborn, M.R. “Transnitrosation and Decomposition of Nitrosamines,” Bogovski, P. and Walker, E.A., Editors, N-nitroso Compounds in the Environment, Lyon, International Agency for Research on Cancer (IARC Scientific Publication No. 9), pp. 94-100 (1974).
7. Burgess, E.M., and Lavanish, J.M. “Photochemical Decomposition of N-nitrosamines,” Tetrahedon Letters, 1221 (1964)
8. Druckrey, H., Preussmann, R., Ivankovic, S., and Schmahl, D. “Organotrope Carcinogene Wirkungen bei 65 Verschiedenen N-NitrosoVerbindungen an BD-Ratten,” Z. Krebsforsch., 69, 103 (1967).
9. Fiddler, W. “The Occurrence and Determination of N-nitroso Compounds,” Toxicol. Appl. Pharmacol., 31, 352 (1975).
10. “Carcinogens - Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
11. “OSHA Safety and Health Standards, General Industry,” (29 CFR Part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
12. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
13. Lijinsky, W. “How Nitrosamines Cause Cancer,” New Scientist, 73, 216 (1977).
14. Mirvish, S.S. “N-Nitroso compounds: Their Chemical and in vivo Formation and Possible Importance as Environmental Carcinogens,” J. Toxicol. Environ. Health, 3, 1267 (1977).
15. “Reconnaissance of Environmental Levels of Nitrosamines in the Central United States,” EPA-330/1-77-001, National Enforcement Investigations Center, U.S. Environmental Protection Agency (1977).
16. “Atmospheric Nitrosamine Assessment Report,” Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency, Research Triangle Park, North Carolina (1976).
17. “Scientific and Technical Assessment Report on Nitrosamines,” EPA-660/6-7-001, Office of Research and Development, U.S. Environmental Protection Agency (1976).
18. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value of 1.22 derived in this report.)
19. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
20. “Methods 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric, DPD) for Chlorine, Total Residual,” Methods for Chemical Analysis of Water and Wastes, EPA-600/4-79-020, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1979.
21. Burke, J. A. “Gas Chromatography for Pesticide Residue Analysis; Some Practical Aspects,” Journal of the Association of Official Analytical Chemists, 48, 1037 (1965).
22. “Method Detection Limit and Analytical Curve Studies EPA Methods 606, 607, and 608,” Special letter report for EPA Contract 68-03-2606, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, June 1980.
23. “EPA Method Study 17 Method 607 - Nitrosamines,” EPA 600/4-84-051, National Technical Information Service, PB84-207646, Springfield, Virginia 22161, June 1984.
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Method detection limit (µg/L) | |
|---|---|---|---|
| Column 1 | Column 2 | ||
| N-Nitrosodimethylamine | 4.1 | 0.88 | 0.15 |
| N-Nitrosodi-n-propylamine | 12.1 | 4.2 | .46 |
| N-Nitrosodiphenylamine a | b 12.8 | c 6.4 | .81 |
Column 1 conditions: Chromosorb W-AW (80/100 mesh) coated with 10% Carbowax 20 M/2% KOH packed in a 1.8 m long × 4mm ID glass column with helium carrier gas at 40 mL/min flow rate. Column temperature held isothermal at 110 °C, except where otherwise indicated.
Column 2 conditions: Supelcoport (100/120 mesh) coated with 10% SP-2250 packed in a 1.8 m long × 4 mm ID glass column with helium carrier gas at 40 mL/min flow rate. Column temperature held isothermal at 120 °C, except where otherwise indicated.
a Measured as diphenylamine.
b 220 °C column temperature.
c 210 °C column temperature.
Table 2 - QC Acceptance Criteria - Method 607
| Parameter | Test conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps (percent) |
|---|---|---|---|---|
| N-Nitrosodimethylamine | 20 | 3.4 | 4.6-20.0 | 13-109 |
| N-Nitrosodiphenyl | 20 | 6.1 | 2.1-24.5 | D-139 |
| N-Nitrosodi-n-propylamine | 20 | 5.7 | 11.5-26.8 | 45-146 |
s = Standard deviation for four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
D = Detected; result must be greater than zero.
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 607
| Parameter | Accuracy, as recovery, X′ (µg/L) | Single analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| N-Nitrosodimethylamine | 0.37C + 0.06 | 0.25X −0.04 | 0.25X + 0.11 |
| N-Nitrosodiphenylamine | 0.64C + 0.52 | 0.36X −1.53 | 0.46X −0.47 |
| N-Nitrosodi-n-propylamine | 0.96C−0.07 | 0.15X + 0.13 | 0.21X + 0.15 |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
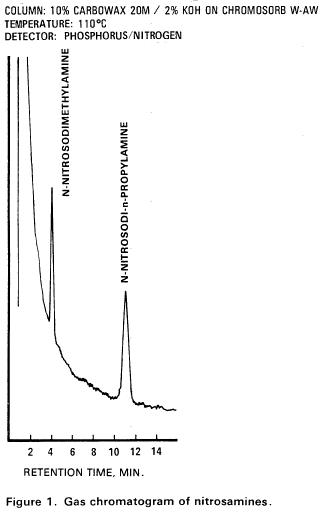
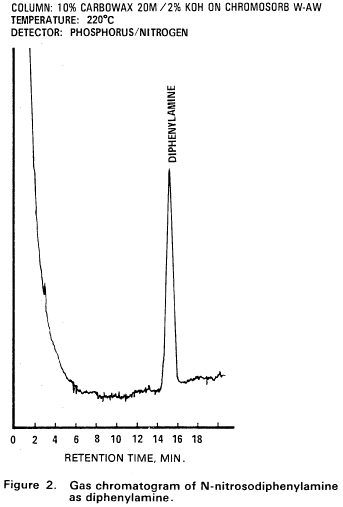 Method 608.3 -
Organochlorine Pesticides And PCBs By GC/HSD 1. Scope and
Application
Method 608.3 -
Organochlorine Pesticides And PCBs By GC/HSD 1. Scope and
Application
1.1 This method is for determination of organochlorine pesticides and polychlorinated biphenyls (PCBs) in industrial discharges and other environmental samples by gas chromatography (GC) combined with a halogen-specific detector (HSD; e.g., electron capture, electrolytic conductivity), as provided under 40 CFR 136.1. This revision is based on a previous protocol (Reference 1), on the revision promulgated October 26, 1984, on an inter-laboratory method validation study (Reference 2), and on EPA Method 1656 (Reference 16). The analytes that may be qualitatively and quantitatively determined using this method and their CAS Registry numbers are listed in Table 1.
1.2 This method may be extended to determine the analytes listed in Table 2. However, extraction or gas chromatography challenges for some of these analytes may make quantitative determination difficult.
1.3 When this method is used to analyze unfamiliar samples for an analyte listed in Table 1 or Table 2, analyte identification must be supported by at least one additional qualitative technique. This method gives analytical conditions for a second GC column that can be used to confirm and quantify measurements. Additionally, Method 625.1 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative confirmation of results for the analytes listed in Tables 1 and 2 using the extract produced by this method, and Method 1699 (Reference 18) provides high resolution GC/MS conditions for qualitative confirmation of results using the original sample. When such methods are used to confirm the identifications of the target analytes, the quantitative results should be derived from the procedure with the calibration range and sensitivity that are most appropriate for the intended application.
1.4 The large number of analytes in Tables 1 and 2 makes testing difficult if all analytes are determined simultaneously. Therefore, it is necessary to determine and perform quality control (QC) tests for the “analytes of interest” only. The analytes of interest are those required to be determined by a regulatory/control authority or in a permit, or by a client. If a list of analytes is not specified, the analytes in Table 1 must be determined, at a minimum, and QC testing must be performed for these analytes. The analytes in Table 1 and some of the analytes in Table 2 have been identified as Toxic Pollutants (40 CFR 401.15), expanded to a list of Priority Pollutants (40 CFR part 423, appendix A).
1.5 In this revision to Method 608, Chlordane has been listed as the alpha- and gamma- isomers in Table 1. Reporting may be by the individual isomers, or as the sum of the concentrations of these isomers, as requested or required by a regulatory/control authority or in a permit. Technical Chlordane is listed in Table 2 and may be used in cases where historical reporting has only been the Technical Chlordane. Toxaphene and the PCBs have been moved from Table 1 to Table 2 (Additional Analytes) to distinguish these analytes from the analytes required in quality control tests (Table 1). QC acceptance criteria for Toxaphene and the PCBs have been retained in Table 4 and may continue to be applied if desired, or if these analytes are requested or required by a regulatory/control authority or in a permit. Method 1668C (Reference 17) may be useful for determination of PCBs as individual chlorinated biphenyl congeners, and Method 1699 (Reference 18) may be useful for determination of the pesticides listed in this method. However, at the time of writing of this revision, Methods 1668C and 1699 had not been approved for use at 40 CFR part 136.
1.6 Method detection limits (MDLs; Reference 3) for the analytes in Tables 1 and some of the analytes in Table 2 are listed in those tables. These MDLs were determined in reagent water (Reference 3). Advances in analytical technology, particularly the use of capillary (open-tubular) columns, allowed laboratories to routinely achieve MDLs for the analytes in this method that are 2-10 times lower than those in the version promulgated in 1984. The MDL for an analyte in a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.6.1 EPA has promulgated this method at 40 CFR part 136 for use in wastewater compliance monitoring under the National Pollutant Discharge Elimination System (NPDES). The data reporting practices described in section 15.6 are focused on such monitoring needs and may not be relevant to other uses of the method.
1.6.2 This method includes “reporting limits” based on EPA's “minimum level” (ML) concept (see the glossary in section 23). Tables 1 and 2 contain MDL values and ML values for many of the analytes.
1.7 The separatory funnel and continuous liquid-liquid sample extraction and concentration steps in this method are essentially the same as those steps in Methods 606, 609, 611, and 612. Thus, a single sample may be extracted to measure the analytes included in the scope of each of these methods. Samples may also be extracted using a disk-based solid-phase extraction (SPE) procedure developed by the 3M Corporation and approved by EPA as an Alternate Test Procedure (ATP) for wastewater analyses in 1995 (Reference 20).
1.8 This method is performance-based. It may be modified to improve performance (e.g., to overcome interferences or improve the accuracy of results) provided all performance requirements are met.
1.8.1 Examples of allowed method modifications are described at 40 CFR 136.6. Other examples of allowed modifications specific to this method are described in section 8.1.2.
1.8.2 Any modification beyond those expressly permitted at 40 CFR 136.6 or in section 8.1.2 of this method shall be considered a major modification subject to application and approval of an alternate test procedure under 40 CFR 136.4 and 136.5.
1.8.3 For regulatory compliance, any modification must be demonstrated to produce results equivalent or superior to results produced by this method when applied to relevant wastewaters (section 8.1.2).
1.9 This method is restricted to use by or under the supervision of analysts experienced in the use of GC/HSD. The laboratory must demonstrate the ability to generate acceptable results with this method using the procedure in section 8.2.
1.10 Terms and units of measure used in this method are given in the glossary at the end of the method.
2. Summary of Method2.1 A measured volume of sample, the amount required to meet an MDL or reporting limit (nominally 1-L), is extracted with methylene chloride using a separatory funnel, a continuous liquid/liquid extractor, or disk-based solid-phase extraction equipment. The extract is dried and concentrated for cleanup, if required. After cleanup, or if cleanup is not required, the extract is exchanged into an appropriate solvent and concentrated to the volume necessary to meet the required compliance or detection limit, and analyzed by GC/HSD.
2.2 Qualitative identification of an analyte in the extract is performed using the retention times on dissimilar GC columns. Quantitative analysis is performed using the peak areas or peak heights for the analyte on the dissimilar columns with either the external or internal standard technique.
2.3 Florisil®, alumina, a C18 solid-phase cleanup, and an elemental sulfur cleanup procedure are provided to aid in elimination of interferences that may be encountered. Other cleanup procedures may be used if demonstrated to be effective for the analytes in a wastewater matrix.
3. Contamination and Interferences3.1 Solvents, reagents, glassware, and other sample processing lab ware may yield artifacts, elevated baselines, or matrix interferences causing misinterpretation of chromatograms. All materials used in the analysis must be demonstrated free from contamination and interferences by running blanks initially and with each extraction batch (samples started through the extraction process in a given 24-hour period, to a maximum of 20 samples - see Glossary for detailed definition), as described in section 8.5. Specific selection of reagents and purification of solvents by distillation in all-glass systems may be required. Where possible, labware is cleaned by extraction or solvent rinse, or baking in a kiln or oven.
3.2 Glassware must be scrupulously cleaned (Reference 4). Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and reagent water. The glassware should then be drained dry, and heated at 400 °C for 15-30 minutes. Some thermally stable materials, such as PCBs, may require higher temperatures and longer baking times for removal. Solvent rinses with pesticide quality acetone, hexane, or other solvents may be substituted for heating. Do not heat volumetric labware above 90 °C. After drying and cooling, store inverted or capped with solvent-rinsed or baked aluminum foil in a clean environment to prevent accumulation of dust or other contaminants.
3.3 Interferences by phthalate esters can pose a major problem in pesticide analysis when using the electron capture detector. The phthalate esters generally appear in the chromatogram as large late eluting peaks, especially in the 15 and 50% fractions from Florisil®. Common flexible plastics contain varying amounts of phthalates that may be extracted or leached from such materials during laboratory operations. Cross contamination of clean glassware routinely occurs when plastics are handled during extraction steps, especially when solvent-wetted surfaces are handled. Interferences from phthalates can best be minimized by avoiding use of non-fluoropolymer plastics in the laboratory. Exhaustive cleanup of reagents and glassware may be required to eliminate background phthalate contamination (References 5 and 6). Interferences from phthalate esters can be avoided by using a microcoulometric or electrolytic conductivity detector.
3.4 Matrix interferences may be caused by contaminants co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. Interferences extracted from samples high in total organic carbon (TOC) may result in elevated baselines, or by enhancing or suppressing a signal at or near the retention time of an analyte of interest. Analyses of the matrix spike and matrix spike duplicate (Section 8.3) may be useful in identifying matrix interferences, and the cleanup procedures in Section 11 may aid in eliminating these interferences. EPA has provided guidance that may aid in overcoming matrix interferences (Reference 7); however, unique samples may require additional cleanup approaches to achieve the MDLs listed in Tables 1 and 2.
4. Safety4.1 Hazards associated with each reagent used in this method have not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of safety data sheets (SDSs, OSHA, 29 CFR 1910.12009(g)) should also be made available to all personnel involved in sample handling and chemical analysis. Additional references to laboratory safety are available and have been identified (References 8 and 9) for the information of the analyst.
4.2 The following analytes covered by this method have been tentatively classified as known or suspected human or mammalian carcinogens: 4,4′-DDT, 4,4′-DDD, the BHCs, and the PCBs. Primary standards of these toxic analytes should be prepared in a chemical fume hood, and a NIOSH/MESA approved toxic gas respirator should be worn when high concentrations are handled.
4.3 This method allows the use of hydrogen as a carrier gas in place of helium (section 5.8.2). The laboratory should take the necessary precautions in dealing with hydrogen, and should limit hydrogen flow at the source to prevent buildup of an explosive mixture of hydrogen in air.
5. Apparatus and Materials Note:Brand names and suppliers are for illustration purposes only. No endorsement is implied. Equivalent performance may be achieved using equipment and materials other than those specified here. Demonstrating that the equipment and supplies used in the laboratory achieve the required performance is the responsibility of the laboratory. Suppliers for equipment and materials in this method may be found through an on-line search. Please do not contact EPA for supplier information.
5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - Amber glass bottle large enough to contain the necessary sample volume (nominally 1 L), fitted with a fluoropolymer-lined screw cap. Foil may be substituted for fluoropolymer if the sample is not corrosive. If amber bottles are not available, protect samples from light. Unless pre-cleaned, the bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must use a glass or fluoropolymer container and tubing for sample collection. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, rinse the compressible tubing thoroughly with methanol, followed by repeated rinsing with reagent water to minimize the potential for sample contamination. An integrating flow meter is required to collect flow proportional composites. The sample container must be kept refrigerated at ≤6 °C and protected from light during compositing.
5.2. Lab ware.
5.2.1 Extraction.
5.2.1.1 pH measurement.
5.2.1.1.1 pH meter, with combination glass electrode.
5.2.1.1.2 pH paper, wide range (Hydrion Papers, or equivalent).
5.2.1.2 Separatory funnel - Size appropriate to hold the sample and extraction solvent volumes, equipped with fluoropolymer stopcock.
5.2.1.3 Continuous liquid-liquid extractor - Equipped with fluoropolymer or glass connecting joints and stopcocks requiring no lubrication. (Hershberg-Wolf Extractor, Ace Glass Company, Vineland, NJ, or equivalent.)
5.2.1.3.1 Round-bottom flask, 500-mL, with heating mantle.
5.2.1.3.2 Condenser, Graham, to fit extractor.
5.2.1.4 Solid-phase extractor - 90-mm filter apparatus (Figure 2) or multi-position manifold.
Note:The approved ATP for solid-phase extraction is limited to disk-based extraction media and associated peripheral equipment.
5.2.1.4.1 Vacuum system - Capable of achieving 0.1 bar (25 inch) Hg (house vacuum, vacuum pump, or water aspirator), equipped with shutoff valve and vacuum gauge.
5.2.1.4.2 Vacuum trap - Made from 500-mL sidearm flask fitted with single-hole rubber stopper and glass tubing.
5.2.2 Filtration.
5.2.2.1 Glass powder funnel, 125- to 250-mL.
5.2.2.2 Filter paper for above, Whatman 41, or equivalent.
5.2.2.3 Prefiltering aids - 90-mm 1-µm glass fiber filter or Empore® Filter Aid 400.
5.2.3 Drying column.
5.2.3.1 Chromatographic column - Approximately 400 mm long x 15 mm ID, with fluoropolymer stopcock and coarse frit filter disc (Kontes or equivalent).
5.2.3.2 Glass wool - Pyrex, extracted with methylene chloride or baked at 450 °C for 1 hour minimum.
5.2.4 Column for Florisil® or alumina cleanup - Approximately 300 mm long x 10 mm ID, with fluoropolymer stopcock. (This column is not required if cartridges containing Florisil® are used.)
5.2.5 Concentration/evaporation.
Note:Use of a solvent recovery system with the K-D or other solvent evaporation apparatus is strongly recommended.
5.2.5.1 Kuderna-Danish concentrator.
5.2.5.1.1 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes or equivalent). Calibration must be checked at the volumes employed for extract volume measurement. A ground-glass stopper is used to prevent evaporation of extracts.
5.2.5.1.2 Evaporative flask, Kuderna-Danish - 500-mL (Kontes or equivalent). Attach to concentrator tube with connectors.
5.2.5.1.3 Snyder column, Kuderna/Danish - Three-ball macro (Kontes or equivalent).
5.2.5.1.4 Snyder column - Two-ball micro (Kontes or equivalent).
5.2.5.1.5 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C), installed in a hood using appropriate engineering controls to limit exposure to solvent vapors.
5.2.5.2 Nitrogen evaporation device - Equipped with heated bath that can be maintained at an appropriate temperature for the solvent and analytes. (N-Evap, Organomation Associates, Inc., or equivalent).
5.2.5.3 Rotary evaporator - Buchi/Brinkman-American Scientific or equivalent, equipped with a variable temperature water bath, vacuum source with shutoff valve at the evaporator, and vacuum gauge.
5.2.5.3.1 A recirculating water pump and chiller are recommended, as use of tap water for cooling the evaporator wastes large volumes of water and can lead to inconsistent performance as water temperatures and pressures vary.
5.2.5.3.2 Round-bottom flask - 100-mL and 500-mL or larger, with ground-glass fitting compatible with the rotary evaporator
Note:This equipment is used to prepare copper foil or copper powder for removing sulfur from sample extracts (see Section 6.7.4).
5.2.5.4 Automated concentrator - Equipped with glassware sufficient to concentrate 3-400 mL extract to a final volume of 1-10 mL under controlled conditions of temperature and nitrogen flow (Turbovap, or equivalent). Follow manufacturer's directions and requirements.
5.2.5.5 Boiling chips - Glass, silicon carbide, or equivalent, approximately 10/40 mesh. Heat at 400 °C for 30 minutes, or solvent rinse or Soxhlet extract with methylene chloride.
5.2.6 Solid-phase extraction disks - 90-mm extraction disks containing 2 g of 8-µm octadecyl (C18) bonded silica uniformly enmeshed in a matrix of inert PTFE fibrils (3M Empore® or equivalent). The disks should not contain any organic compounds, either from the PTFE or the bonded silica, which will leach into the methylene chloride eluant. One liter of reagent water should pass through the disks in 2-5 minutes, using a vacuum of at least 25 inches of mercury.
Note:Extraction disks from other manufacturers may be used in this procedure, provided that they use the same solid-phase materials (i.e., octadecyl bonded silica). Disks of other diameters also may be used, but may adversely affect the flow rate of the sample through the disk.
5.3 Vials.
5.3.1 Extract storage - 10- to 15-mL, amber glass, with fluoropolymer-lined screw cap.
5.3.2 GC autosampler - 1- to 5-mL, amber glass, with fluoropolymer-lined screw- or crimp-cap, to fit GC autosampler.
5.4 Balances.
5.4.1 Analytical - Capable of accurately weighing 0.1 mg.
5.4.2 Top loading - Capable of weighing 10 mg.
5.5 Sample cleanup.
5.5.1 Oven - For baking and storage of adsorbents, capable of maintaining a constant temperature (±5 °C) in the range of 105-250 °C.
5.5.2 Muffle furnace - Capable of cleaning glassware or baking sodium sulfate in the range of 400-450 °C.
5.5.3 Vacuum system and cartridges for solid-phase cleanup (see Section 11.2).
5.5.3.1 Vacuum system - Capable of achieving 0.1 bar (25 in.) Hg (house vacuum, vacuum pump, or water aspirator), equipped with shutoff valve and vacuum gauge.
5.5.3.2 VacElute Manifold (Analytichem International, or equivalent).
5.5.3.3 Vacuum trap - Made from 500-mL sidearm flask fitted with single-hole rubber stopper and glass tubing.
5.5.3.4 Rack for holding 50-mL volumetric flasks in the manifold.
5.5.3.5 Cartridge - Mega Bond Elute, Non-polar, C18 Octadecyl, 10 g/60 mL (Analytichem International or equivalent), used for solid-phase cleanup of sample extracts (see Section 11.2).
5.5.4 Sulfur removal tube - 40- to 50-mL bottle, test tube, or Erlenmeyer flask with fluoropolymer-lined screw cap.
5.6 Centrifuge apparatus.
5.6.1 Centrifuge - Capable of rotating 500-mL centrifuge bottles or 15-mL centrifuge tubes at 5,000 rpm minimum.
5.6.2 Centrifuge bottle - 500-mL, with screw cap, to fit centrifuge.
5.6.3 Centrifuge tube - 15-mL, with screw cap, to fit centrifuge.
5.7 Miscellaneous lab ware - Graduated cylinders, pipettes, beakers, volumetric flasks, vials, syringes, and other lab ware necessary to support the operations in this method.
5.8 Gas chromatograph - Dual-column with simultaneous split/splitless, temperature programmable split/splitless (PTV), or on-column injection; temperature program with isothermal holds, and all required accessories including syringes, analytical columns, gases, and detectors. An autosampler is highly recommended because it injects volumes more reproducibly than manual injection techniques. Alternatively, two separate single-column gas chromatographic systems may be employed.
5.8.1 Example columns and operating conditions.
5.8.1.1 DB-608 (or equivalent), 30-m long x 0.53-mm ID fused-silica capillary, 0.83-µm film thickness.
5.8.1.2 DB-1701 (or equivalent), 30-m long x 0.53-mm ID fused-silica capillary, 1.0-µm film thickness.
5.8.1.3 Suggested operating conditions used to meet the retention times shown in Table 3 are:
(a) Carrier gas flow rate: Approximately 7 mL/min,
(b) Initial temperature: 150 °C for 0.5 minute,
(c) Temperature program: 150-270 °C at 5 °C/min, and
(d) Final temperature: 270 °C, until trans-Permethrin elutes.
Note:Other columns, internal diameters, film thicknesses, and operating conditions may be used, provided that the performance requirements in this method are met. However, the column pair chosen must have dissimilar phases/chemical properties in order to separate the compounds of interest in different retention time order. Columns that only differ in the length, ID, or film thickness, but use the same stationary phase do not qualify as “dissimilar.”
5.8.2 Carrier gas - Helium or hydrogen. Data in the tables in this method were obtained using helium carrier gas. If hydrogen is used, analytical conditions may need to be adjusted for optimum performance, and calibration and all QC tests must be performed with hydrogen carrier gas. See Section 4.3 for precautions regarding the use of hydrogen as a carrier gas.
5.8.3 Detector - Halogen-specific detector (electron capture detector [ECD], electrolytic conductivity detector [ELCD], or equivalent). The ECD has proven effective in the analysis of wastewaters for the analytes listed in Tables 1 and 2, and was used to develop the method performance data in Section 17 and Tables 4 and 5.
5.8.4 Data system - A computer system must be interfaced to the GC that allows continuous acquisition and storage of data from the detectors throughout the chromatographic program. The computer must have software that allows searching GC data for specific analytes, and for plotting responses versus time. Software must also be available that allows integrating peak areas or peak heights in selected retention time windows and calculating concentrations of the analytes.
6. Reagents and Standards6.1 pH adjustment.
6.1.1 Sodium hydroxide solutions.
6.1.1.1 Concentrated (10 M) - Dissolve 40 g of NaOH (ACS) in reagent water and dilute to 100 mL.
6.1.1.2 Dilute (1 M) - Dissolve 40 g NaOH in 1 L of reagent water.
6.1.2 Sulfuric acid (1+1) - Slowly add 50 mL of H2SO4 (ACS, sp. gr. 1.84) to 50 mL of reagent water.
6.1.3 Hydrochloric acid - Reagent grade, 6 N.
6.2 Sodium thiosulfate - (ACS) granular.
6.3 Sodium sulfate - Sodium sulfate, reagent grade, granular anhydrous (Baker or equivalent), rinsed with methylene chloride, baked in a shallow tray at 450 °C for 1 hour minimum, cooled in a desiccator, and stored in a pre-cleaned glass bottle with screw cap which prevents moisture from entering. If, after heating, the sodium sulfate develops a noticeable grayish cast (due to the presence of carbon in the crystal matrix), that batch of reagent is not suitable for use and should be discarded. Extraction with methylene chloride (as opposed to simple rinsing) and baking at a lower temperature may produce sodium sulfate suitable for use.
6.4 Reagent water - Reagent water is defined as water in which the analytes of interest and interfering compounds are not observed at the MDLs of the analytes in this method.
6.5 Solvents - Methylene chloride, acetone, methanol, hexane, acetonitrile, and isooctane, high purity pesticide quality, or equivalent, demonstrated to be free of the analytes and interferences (section 3). Purification of solvents by distillation in all-glass systems may be required.
Note:The standards and final sample extracts must be prepared in the same final solvent.
6.6 Ethyl ether - Nanograde, redistilled in glass if necessary. Ethyl ether must be shown to be free of peroxides before use, as indicated by EM Laboratories Quant test strips (available from Scientific Products Co. and other suppliers). Procedures recommended for removal of peroxides are provided with the test strips. After removal of peroxides, add 20 mL of ethyl alcohol preservative to each liter of ether.
6.7 Materials for sample cleanup.
6.7.1 Florisil® - PR grade (60/100 mesh), activated at 650-700 °C, stored in the dark in a glass container with fluoropolymer-lined screw cap. Activate each batch immediately prior to use for 16 hours minimum at 130 °C in a foil-covered glass container and allow to cool. Alternatively, 500 mg cartridges (J.T. Baker, or equivalent) may be used.
6.7.1.1 Cartridge certification - Each cartridge lot must be certified to ensure recovery of the analytes of interest and removal of 2,4,6-trichlorophenol. To make the test mixture, add the trichlorophenol solution (section 6.7.1.3) to the same standard used to prepare the Quality Control Check Sample (section 6.8.3). Transfer the mixture to the column and dry the column. Pre-elute with three 10-mL portions of elution solvent, drying the column between elutions. Elute the cartridge with 10 mL each of methanol and water, as in section 11.2.3.3.
6.7.1.2 Concentrate the eluant to per section 10.3.3, exchange to isooctane or hexane per section 10.3.3, and inject 1.0 µL of the concentrated eluant into the GC using the procedure in section 12. The recovery of all analytes (including the unresolved GC peaks) shall be within the ranges for calibration verification (section 13.6 and Table 4), the recovery of trichlorophenol shall be less than 5%, and no peaks interfering with the target analytes shall be detected. Otherwise the Florisil cartridge is not performing properly and the cartridge lot shall be rejected.
6.7.1.3 Florisil cartridge calibration solution - 2,4,6-Trichlorophenol, 0.1 µg/mL in acetone.
6.7.2 SPE elution solvent - Methylene chloride:acetonitrile:hexane (50:3:47).
6.7.3 Alumina, neutral, Brockman Activity I, 80-200 mesh (Fisher Scientific certified, or equivalent). Heat in a glass bottle for 16 hours at 400 to 450 °C. Seal and cool to room temperature. Add 7% (w/w) reagent water and mix for 10 to 12 hours. Keep bottle tightly sealed.
6.7.4 Sulfur removal.
6.7.4.1 Copper foil or powder - Fisher, Alfa Aesar, or equivalent. Cut copper foil into approximately 1-cm squares. Copper must be activated before it may be used, as described below.
6.7.4.1.1 Place the quantity of copper needed for sulfur removal (section 11.5.1.3) in a ground-glass-stoppered Erlenmeyer flask or bottle. Cover the foil or powder with methanol.
6.7.4.1.2 Add HCl dropwise (0.5-1.0 mL) while swirling, until the copper brightens.
6.7.4.1.3 Pour off the methanol/HCl and rinse 3 times with reagent water to remove all traces of acid, then 3 times with acetone, then 3 times with hexane.
6.7.4.1.4 For copper foil, cover with hexane after the final rinse. Store in a stoppered flask under nitrogen until used. For the powder, dry on a rotary evaporator. Store in a stoppered flask under nitrogen until used. Inspect the copper foil or powder before each use. It must have a bright, non-oxidized appearance to be effective. Copper foil or powder that has oxidized may be reactivated using the procedure described above.
6.7.4.2 Tetrabutylammonium sulfite (TBA sulfite) - Prepare as described below.
6.7.4.2.1 Tetrabutylammonium hydrogen sulfate, [CH3(CH2)3]4NHSO4.
6.7.4.2.2 Sodium sulfite, Na2SO3.
6.7.4.2.3 Dissolve approximately 3 g tetrabutylammonium hydrogen sulfate in 100 mL of reagent water in an amber bottle with fluoropolymer-lined screw cap. Extract with three 20-mL portions of hexane and discard the hexane extracts.
6.7.4.2.4 Add 25 g sodium sulfite to produce a saturated solution. Store at room temperature. Replace after 1 month.
6.7.5 Sodium chloride - Reagent grade, prepare at 5% (w/v) solution in reagent water.
6.8 Stock standard solutions - Stock standard solutions may be prepared from pure materials, or purchased as certified solutions. Traceability must be to the National Institute of Standards and Technology (NIST) or other national or international standard, when available. Stock solution concentrations alternative to those below may be used. Because of the toxicity of some of the compounds, primary dilutions should be prepared in a hood, and a NIOSH/MESA approved toxic gas respirator should be worn when high concentrations of neat materials are handled. The following procedure may be used to prepare standards from neat materials.
6.8.1 Accurately weigh about 0.0100 g of pure material in a 10-mL volumetric flask. Dilute to volume in pesticide quality hexane, isooctane, or other suitable solvent. Larger volumes may be used at the convenience of the laboratory. When compound purity is assayed to be 96% or greater, the weight may be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards may be used at any concentration if they are certified by the manufacturer or by an independent source.
6.8.1.1 Unless stated otherwise in this method, store non-aqueous standards in fluoropolymer-lined screw-cap, or heat-sealed, glass containers, in the dark at −20 to −10 °C. Store aqueous standards; e.g., the aqueous LCS (section 8.4), in the dark at ≤6 °C, but do not freeze.
6.8.1.2 Standards prepared by the laboratory may be stored for up to one year, except when comparison with QC check standards indicates that a standard has degraded or become more concentrated due to evaporation, or unless the laboratory has data on file to prove stability for a longer period. Commercially prepared standards may be stored until the expiration date provided by the vendor, except when comparison with QC check standards indicates that a standard has degraded or become more concentrated due to evaporation, or unless the laboratory has data from the vendor on file to prove stability for a longer period.
6.8.2 Calibration solutions - It is necessary to prepare calibration solutions for the analytes of interest (section 1.4) only using an appropriate solvent (isooctane or hexane may be used). Whatever solvent is used, both the calibration standards and the final sample extracts must use the same solvent. Other analytes may be included as desired.
6.8.2.1 Prepare calibration standards for the single-component analytes of interest and surrogates at a minimum of three concentration levels (five are suggested) by adding appropriate volumes of one or more stock standards to volumetric flasks. One of the calibration standards should be at a concentration at or below the ML specified in Table 1, or 2, or as specified by a regulatory/control authority or in a permit. The ML value may be rounded to a whole number that is more convenient for preparing the standard, but must not exceed the ML value listed in Tables 1 or 2 for those analytes which list ML values. Alternatively, the laboratory may establish an ML for each analyte based on the concentration of the lowest calibration standard in a series of standards produced by the laboratory or obtained from a commercial vendor, again, provided that the ML does not exceed the ML in Table 1 and 2, and provided that the resulting calibration meets the acceptance criteria in section 7.5.2 based on the RSD, RSE, or R 2.
(a) The other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the GC system. A minimum of six concentration levels is required for a second order, non-linear (e.g., quadratic; ax 2 + bx + c = 0) calibration (section 7.5.2 or 7.6.2). Calibrations higher than second order are not allowed. A separate standard near the MDL may be analyzed as a check on sensitivity, but should not be included in the linearity assessment. The solvent for the standards must match the final solvent for the sample extracts (e.g., isooctane or hexane).
Note:The option for non-linear calibration may be necessary to address specific instrumental techniques. However, it is not EPA's intent to allow non-linear calibration to be used to compensate for detector saturation or to avoid proper instrument maintenance.
(b) Given the number of analytes included in this method, it is highly likely that some will coelute on one or both of the GC columns used for the analysis. Divide the analytes into two or more groups and prepare separate calibration standards for each group, at multiple concentrations (e.g., a five-point calibration will require ten solutions to cover two groups of analytes). Table 7 provides information on dividing the target analytes into separate calibration mixtures that should minimize or eliminate co-elutions. This table is provided solely as guidance, based on the GC columns suggested in this method. If an analyte listed in Table 7 is not an analyte of interest in a given laboratory setting, then it need not be included in a calibration mixture.
Note:Many commercially available standards are divided into separate mixtures to address this issue.
(c) If co-elutions occur in analysis of a sample, a co-elution on one column is acceptable so long as effective separation of the co-eluting compounds can be achieved on the second column.
6.8.2.2 Multi-component analytes (e.g., PCBs as Aroclors, and Toxaphene).
6.8.2.2.1 A standard containing a mixture of Aroclor 1016 and Aroclor 1260 will include many of the peaks represented in the other Aroclor mixtures. As a result, a multi-point initial calibration employing a mixture of Aroclors 1016 and 1260 at three to five concentrations should be sufficient to demonstrate the linearity of the detector response without the necessity of performing multi-point initial calibrations for each of the seven Aroclors. In addition, such a mixture can be used as a standard to demonstrate that a sample does not contain peaks that represent any one of the Aroclors. This standard can also be used to determine the concentrations of either Aroclor 1016 or Aroclor 1260, should they be present in a sample. Therefore, prepare a minimum of three calibration standards containing equal concentrations of both Aroclor 1016 and Aroclor 1260 by dilution of the stock standard with isooctane or hexane. The concentrations should correspond to the expected range of concentrations found in real samples and should bracket the linear range of the detector.
6.8.2.2.2 Single standards of each of the other five Aroclors are required to aid the analyst in pattern recognition. Assuming that the Aroclor 1016/1260 standards described in Section 6.8.2.2.1 have been used to demonstrate the linearity of the detector, these single standards of the remaining five Aroclors also may be used to determine the calibration factor for each Aroclor. Prepare a standard for each of the other Aroclors. The concentrations should generally correspond to the mid-point of the linear range of the detector, but lower concentrations may be employed at the discretion of the analyst based on project requirements.
6.8.2.2.3 For Toxaphene, prepare a minimum of three calibration standards containing Toxaphene by dilution of the stock standard with isooctane or hexane. The concentrations should correspond to the expected range of concentrations found in real samples and should bracket the linear range of the detector.
6.8.3 Quality Control (QC) Check Sample Concentrate - Prepare one or more mid-level standard mixtures (concentrates) in acetone (or other water miscible solvent). The concentrate is used as the spiking solution with which to prepare the Demonstration of Capabilities (DOC) samples, the Laboratory Control Sample (LCS), and Matrix Spike (MS) and Matrix Spike Duplicate (MSD) samples described in section 8. If prepared by the laboratory (as opposed the purchasing it from a commercial supplier), the concentrate must be prepared independently from the standards used for calibration, but may be prepared from the same source as the second-source standard used for calibration verification (section 7.7). Regardless of the source, the concentrate must be in a water-miscible solvent, as noted above. The concentrate is used to prepare the DOC and LCS (sections 8.2.1 and 8.4) and MS/MSD samples (section 8.3). Depending on the analytes of interest for a given sample (see Section 1.4), multiple solutions and multiple LCS or MS/MSD samples may be required to account for co-eluting analytes. However, a co-elution on one column is acceptable so long as effective separation of the co-eluting compounds can be achieved on the second column. In addition, the concentrations of the MS/MSD samples should reflect any relevant compliance limits for the analytes of interest, as described in section 8.3.1. If a custom spiking solution is required for a specific discharge (section 8.3.1), prepare it separately from the DOC and LCS solution.
Note:Some commercially available standards are divided into separate mixtures to address the co-elution issue.
6.8.4 Calibration Verification Standards - In order to verify the results of the initial calibration standards, prepare one or more mid-level standard mixtures in isooctane or hexane, using standards obtained from a second source (different manufacturer or different certified lot from the calibration standards). These standards will be analyzed to verify the accuracy of the calibration (sections 7.7 and 13.6.2). As with the QC sample concentrate in section 6.8.3, multiple solutions may be required to address co-elutions among all of the analytes.
6.8.5 Internal standard solution - If the internal standard calibration technique is to be used, prepare pentachloronitrobenzene (PCNB) at a concentration of 10 µg/mL in ethyl acetate. Alternative and multiple internal standards; e.g., tetrachloro-m-xylene, 4,4′-dibromobiphenyl, and/or decachlorobiphenyl may be used provided that the laboratory performs all QC tests and meets all QC acceptance criteria with the alternative or additional internal standard(s) as an integral part of this method.
6.8.6 Surrogate solution - Prepare a solution containing one or more surrogates at a concentration of 2 µg/mL in acetone. Potential surrogates include: dibutyl chlorendate (DBC), tetrachloro-m-xylene (TCMX), 4,4′-dibromobiphenyl, or decachlorobiphenyl. Alternative surrogates and concentrations may be used, provided the laboratory performs all QC tests and meets all QC acceptance criteria with the alternative surrogate(s) as an integral part of this method. If the internal standard calibration technique is used, do not use the internal standard as a surrogate.
6.8.7 DDT and endrin decomposition (breakdown) solution - Prepare a solution containing endrin at a concentration of 50 ng/mL and 4,4'-DDT at a concentration of 100 ng/mL, in isooctane or hexane. A 1-µL injection of this standard will contain 50 picograms (pg) of endrin and 100 pg of DDT. The concentration of the solution may be adjusted by the laboratory to accommodate other injection volumes such that the same masses of the two analytes are introduced into the instrument.
7. Calibration7.1 Establish gas chromatographic operating conditions equivalent to those in Section 5.8.1 and Footnote 2 to Table 3. Alternative temperature program and flow rate conditions may be used. The system may be calibrated using the external standard technique (section 7.5) or the internal standard technique (section 7.6). It is necessary to calibrate the system for the analytes of interest (section 1.4) only.
7.2 Separately inject the mid-level calibration standard for each calibration mixture. Store the retention time on each GC column.
7.3 Injection of calibration solutions - Inject a constant volume in the range of 0.5 to 2.0 µL of each calibration solution into the GC column/detector pairs. An alternative volume (see Section 12.3) may be used provided all requirements in this method are met. Beginning with the lowest level mixture and proceeding to the highest level mixture may limit the risk of carryover from one standard to the next, but other sequences may be used. An instrument blank should be analyzed after the highest standard to demonstrate that there is no carry-over within the system for this calibration range.
7.4 For each analyte, compute, record, and store, as a function of the concentration injected, the retention time and peak area on each column/detector system. If multi-component analytes are to be analyzed, store the retention time and peak area for the three to five exclusive (unique large) peaks for each PCB or technical chlordane. Use four to six peaks for toxaphene.
7.5 External standard calibration.
7.5.1 From the calibration data (Section 7.4), calculate the calibration factor (CF) for each analyte at each concentration according to the following equation:
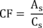 Where: Cs
= Concentration of the analyte in the standard (ng/mL) As = Peak
height or area
Where: Cs
= Concentration of the analyte in the standard (ng/mL) As = Peak
height or area
For multi-component analytes, choose a series of characteristic peaks for each analyte (3 to 5 for each Aroclor, 4 to 6 for toxaphene) and calculate individual calibration factors for each peak. Alternatively, for toxaphene, sum the areas of all of the peaks in the standard chromatogram and use the summed area to determine the calibration factor. (If this alternative is used, the same approach must be used to quantitate the analyte in the samples.)
7.5.2 Calculate the mean (average) and relative standard deviation (RSD) of the calibration factors. If the RSD is less than 20%, linearity through the origin can be assumed and the average CF can be used for calculations. Alternatively, the results can be used to fit a linear or quadratic regression of response, As, vs. concentration Cs. If used, the regression must be weighted inversely proportional to concentration. The coefficient of determination (R 2) of the weighted regression must be greater than 0.920. Alternatively, the relative standard error (Reference 10) may be used as an acceptance criterion. As with the RSD, the RSE must be less than 20%. If an RSE less than 20% cannot be achieved for a quadratic regression, system performance is unacceptable and the system must be adjusted and re-calibrated.
Note:Regression calculations are not included in this method because the calculations are cumbersome and because many GC/ECD data systems allow selection of weighted regression for calibration and calculation of analyte concentrations.
7.6 Internal standard calibration.
7.6.1 From the calibration data (Section 7.4), calculate the response factor (RF) for each analyte at each concentration according to the following equation:
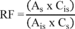 Where: As
= Response for the analyte to be measured. Ais = Response for the
internal standard. Cis = Concentration of the internal standard
(ng/mL) Cs = Concentration of the analyte to be measured (ng/mL).
Where: As
= Response for the analyte to be measured. Ais = Response for the
internal standard. Cis = Concentration of the internal standard
(ng/mL) Cs = Concentration of the analyte to be measured (ng/mL).
7.6.2 Calculate the mean (average) and relative standard deviation (RSD) of the response factors. If the RSD is less than 15%, linearity through the origin can be assumed and the average RF can be used for calculations. Alternatively, the results can be used to prepare a calibration curve of response ratios, As/Ais, vs. concentration ratios, Cs/Cis, for the analyte. A minimum of six concentration levels is required for a non-linear (e.g., quadratic) regression. If used, the regression must be weighted inversely proportional to concentration, and the coefficient of determination of the weighted regression must be greater than 0.920. Alternatively, the relative standard error (Reference 10) may be used as an acceptance criterion. As with the RSD, the RSE must be less than 15%. If an RSE less than 15% cannot be achieved for a quadratic regression, system performance is unacceptable and the system must be adjusted and re-calibrated.
7.7 The working calibration curve, CF, or RF must be verified immediately after calibration and at the beginning and end of each 24-hour shift by the analysis of a mid-level calibration standard. The calibration verification standard(s) must be obtained from a second manufacturer or a manufacturer's batch prepared independently from the batch used for calibration (Section 6.8.4). Requirements for calibration verification are given in Section 13.6 and Table 4. Alternatively, calibration verification may be performed after a set number of injections (e.g., every 20 injections), to include injection of extracts of field samples, QC samples, instrument blanks, etc. (i.e., it is based on the number of injections performed, not sample extracts). The time for the injections may not exceed 24 hours.
Note:The 24-hour shift begins after analysis of the combined QC standard (calibration verification) and ends 24 hours later. The ending calibration verification standard is run immediately after the last sample run during the 24-hour shift, so the beginning and ending calibration verifications are outside of the 24-hour shift. If calibration verification is based on the number of injections instead of time, then the ending verification standard for one group of injections may be used as the beginning verification for the next group of injections.
7.8 Florisil® calibration - The column cleanup procedure in Section 11.3 utilizes Florisil column chromatography. Florisil® from different batches or sources may vary in adsorptive capacity. To standardize the amount of Florisil® which is used, use of the lauric acid value (Reference 11) is suggested. The referenced procedure determines the adsorption from a hexane solution of lauric acid (mg) per g of Florisil®. The amount of Florisil® to be used for each column is calculated by dividing 110 by this ratio and multiplying by 20 g. If cartridges containing Florisil® are used, then this step is not necessary.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality assurance program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and ongoing analysis of spiked samples and blanks to evaluate and document data quality. The laboratory must maintain records to document the quality of data generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet performance requirements of this method. A quality control check standard (LCS, section 8.4) must be prepared and analyzed with each batch of samples to confirm that the measurements were performed in an in-control mode of operation. A laboratory may develop its own performance criteria (as QC acceptance criteria), provided such criteria are as or more restrictive than the criteria in this method.
8.1.1 The laboratory must make an initial demonstration of the capability (IDC) to generate acceptable precision and recovery with this method. This demonstration is detailed in Section 8.2. On a continuing basis, the laboratory must repeat demonstration of capability (DOC) at least annually.
8.1.2 In recognition of advances that are occurring in analytical technology, and to overcome matrix interferences, the laboratory is permitted certain options (section 1.8 and 40 CFR 136.6(b) [Reference 12]) to improve separations or lower the costs of measurements. These options may include alternative extraction (e.g., other solid-phase extraction materials and formats), concentration, and cleanup procedures, and changes in GC columns (Reference 12). Alternative determinative techniques, such as the substitution of spectroscopic or immunoassay techniques, and changes that degrade method performance, are not allowed. If an analytical technique other than the techniques specified in this method is used, that technique must have a specificity equal to or greater than the specificity of the techniques in this method for the analytes of interest. The laboratory is also encouraged to participate in performance evaluation studies (see section 8.8).
8.1.2.1 Each time a modification listed above is made to this method, the laboratory is required to repeat the procedure in section 8.2. If the detection limit of the method will be affected by the change, the laboratory is required to demonstrate that the MDLs (40 CFR part 136, appendix B) are lower than one-third the regulatory compliance limit or as low as the MDLs in this method, whichever are greater. If calibration will be affected by the change, the instrument must be recalibrated per section 7. Once the modification is demonstrated to produce results equivalent or superior to results produced by this method as written, that modification may be used routinely thereafter, so long as the other requirements in this method are met (e.g., matrix spike/matrix spike duplicate recovery and relative percent difference).
8.1.2.1.1 If an allowed method modification, is to be applied to a specific discharge, the laboratory must prepare and analyze matrix spike/matrix spike duplicate (MS/MSD) samples (section 8.3) and LCS samples (section 8.4). The laboratory must include surrogates (Section 8.7) in each of the samples. The MS/MSD and LCS samples must be fortified with the analytes of interest (section 1.4). If the modification is for nationwide use, MS/MSD samples must be prepared from a minimum of nine different discharges (See section 8.1.2.1.2), and all QC acceptance criteria in this method must be met. This evaluation only needs to be performed once other than for the routine QC required by this method (for example it could be performed by the vendor of an alternative material) but any laboratory using that specific material must have the results of the study available. This includes a full data package with the raw data that will allow an independent reviewer to verify each determination and calculation performed by the laboratory (see section 8.1.2.2.5, items (a)-(q)).
8.1.2.1.2 Sample matrices on which MS/MSD tests must be performed for nationwide use of an allowed modification:
(a) Effluent from a publicly owned treatment works (POTW).
(b) ASTM D5905 Standard Specification for Substitute Wastewater.
(c) Sewage sludge, if sewage sludge will be in the permit.
(d) ASTM D1141 Standard Specification for Substitute Ocean Water, if ocean water will be in the permit.
(e) Untreated and treated wastewaters up to a total of nine matrix types (see https://www.epa.gov/eg/industrial-effluent-guidelines for a list of industrial categories with existing effluent guidelines).
(i) At least one of the above wastewater matrix types must have at least one of the following characteristics:
(A) Total suspended solids greater than 40 mg/L.
(B) Total dissolved solids greater than 100 mg/L.
(C) Oil and grease greater than 20 mg/L.
(D) NaCl greater than 120 mg/L.
(E) CaCO3 greater than 140 mg/L.
(ii) The interim acceptance criteria for MS, MSD recoveries that do not have recovery limits in Table 4 or developed in section 8.3.3, and for surrogates that do not have recovery limits developed in section 8.6, must be no wider than 60-140%, and the relative percent difference (RPD) of the concentrations in the MS and MSD that do not have RPD limits in Table 4 or developed in section 8.3.3, must be less than 30%. Alternatively, the laboratory may use the laboratory's in-house limits if they are tighter.
(f) A proficiency testing (PT) sample from a recognized provider, in addition to tests of the nine matrices (section 8.1.2.1.1).
8.1.2.2 The laboratory must maintain records of modifications made to this method. These records include the following, at a minimum:
8.1.2.2.1 The names, titles, and business street addresses, telephone numbers, and email addresses, of the analyst(s) that performed the analyses and modification, and of the quality control officer that witnessed and will verify the analyses and modifications.
8.1.2.2.2 A list of analytes, by name and CAS Registry number.
8.1.2.2.3 A narrative stating reason(s) for the modifications.
8.1.2.2.4 Results from all quality control (QC) tests comparing the modified method to this method, including:
(a) Calibration (section 7).
(b) Calibration verification (section 13.6).
(c) Initial demonstration of capability (section 8.2).
(d) Analysis of blanks (section 8.5).
(e) Matrix spike/matrix spike duplicate analysis (section 8.3).
(f) Laboratory control sample analysis (section 8.4).
8.1.2.2.5 Data that will allow an independent reviewer to validate each determination by tracing the instrument output (peak height, area, or other signal) to the final result. These data are to include:
(a) Sample numbers and other identifiers.
(b) Extraction dates.
(c) Analysis dates and times.
(d) Analysis sequence/run chronology.
(e) Sample weight or volume (section 10).
(f) Extract volume prior to each cleanup step (sections 10 and 11).
(g) Extract volume after each cleanup step (section 11).
(h) Final extract volume prior to injection (sections 10 and 12).
(i) Injection volume (sections 12.3 and 13.2).
(j) Sample or extract dilution (section 15.4).
(k) Instrument and operating conditions.
(l) Column (dimensions, material, etc.).
(m) Operating conditions (temperatures, flow rates, etc.).
(n) Detector (type, operating conditions, etc.).
(o) Chromatograms and other recordings of raw data.
(p) Quantitation reports, data system outputs, and other data to link the raw data to the results reported.
(q) A written Standard Operating Procedure (SOP).
8.1.2.2.6 Each individual laboratory wishing to use a given modification must perform the start-up tests in section 8.1.2 (e.g., DOC, MDL), with the modification as an integral part of this method prior to applying the modification to specific discharges. Results of the DOC must meet the QC acceptance criteria in Table 5 for the analytes of interest (section 1.4), and the MDLs must be equal to or lower than the MDLs in Tables 1 and 2 for the analytes of interest.
8.1.3 Before analyzing samples, the laboratory must analyze a blank to demonstrate that interferences from the analytical system, lab ware, and reagents, are under control. Each time a batch of samples is extracted or reagents are changed, a blank must be extracted and analyzed as a safeguard against laboratory contamination. Requirements for the blank are given in section 8.5.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze samples to monitor and evaluate method and laboratory performance on the sample matrix. The procedure for spiking and analysis is given in section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through analysis of a quality control check sample (laboratory control sample, LCS; on-going precision and recovery sample, OPR) that the measurement system is in control. This procedure is described in Section 8.4.
8.1.6 The laboratory should maintain performance records to document the quality of data that is generated. This procedure is given in section 8.7.
8.1.7 The large number of analytes tested in performance tests in this method present a substantial probability that one or more will fail acceptance criteria when all analytes are tested simultaneously, and a re-test (reanalysis) is allowed if this situation should occur. If, however, continued re-testing results in further repeated failures, the laboratory should document the failures and either avoid reporting results for the analytes that failed or report the problem and failures with the data. A QC failure does not relieve a discharger or permittee of reporting timely results.
8.2 Demonstration of capability (DOC) - To establish the ability to generate acceptable recovery and precision, the laboratory must perform the DOC in sections 8.2.1 through 8.2.6 for the analytes of interest initially and in an on-going manner at least annually. The laboratory must also establish MDLs for the analytes of interest using the MDL procedure at 40 CFR part 136, appendix B. The laboratory's MDLs must be equal to or lower than those listed in Tables 1 or 2, or lower than one-third the regulatory compliance limit, whichever is greater. For MDLs not listed in Tables 1 or 2, the laboratory must determine the MDLs using the MDL procedure at 40 CFR part 136, appendix B under the same conditions used to determine the MDLs for the analytes listed in Tables 1 and 2. When analyzing the PCBs as Aroclors, it is only necessary to establish an MDL for one of the multi-component analytes (e.g., PCB 1254), or the mixture of Aroclors 1016 and 1260 may be used to establish MDLs for all of the Aroclors. Similarly, MDLs for other multi-component analytes (e.g., Chlordanes) may be determined using only one of the major components. All procedures used in the analysis, including cleanup procedures, must be included in the DOC.
8.2.1 For the DOC, a QC check sample concentrate containing each analyte of interest (section 1.4) is prepared in a water-miscible solvent using the solution in section 6.8.3.
Note:QC check sample concentrates are no longer available from EPA.
8.2.2 Using a pipet or syringe, prepare four QC check samples by adding an appropriate volume of the concentrate and of the surrogate(s) to each of four 1-L aliquots of reagent water. Swirl or stir to mix.
8.2.3 Extract and analyze the well-mixed QC check samples according to the method beginning in section 10.
8.2.4 Calculate the average percent recovery (X) and the standard deviation (s) of the percent recovery for each analyte using the four results.
8.2.5 For each analyte, compare s and X with the corresponding acceptance criteria for precision and recovery in Table 4. For analytes in Table 2 that are not listed in Table 4, QC acceptance criteria must be developed by the laboratory. EPA has provided guidance for development of QC acceptance criteria (References 12 and 13). If s and X for all analytes of interest meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for recovery, system performance is unacceptable for that analyte.
Note:The large number of analytes in Tables 1 and 2 present a substantial probability that one or more will fail at least one of the acceptance criteria when many or all analytes are determined simultaneously.
8.2.6 When one or more of the analytes tested fail at least one of the acceptance criteria, repeat the test for only the analytes that failed. If results for these analytes pass, system performance is acceptable and analysis of samples and blanks may proceed. If one or more of the analytes again fail, system performance is unacceptable for the analytes that failed the acceptance criteria. Correct the problem and repeat the test (section 8.2). See section 8.1.7 for disposition of repeated failures.
Note:To maintain the validity of the test and re-test, system maintenance and/or adjustment is not permitted between this pair of tests.
8.3 Matrix spike and matrix spike duplicate (MS/MSD) - The purpose of the MS/MSD requirement is to provide data that demonstrate the effectiveness of the method as applied to the samples in question by a given laboratory, and both the data user (discharger, permittee, regulated entity, regulatory/control authority, customer, other) and the laboratory share responsibility for provision of such data. The data user should identify the sample and the analytes of interest (section 1.4) to be spiked and provide sufficient sample volume to perform MS/MSD analyses. The laboratory must, on an ongoing basis, spike at least 5% of the samples in duplicate from each discharge being monitored to assess accuracy (recovery and precision). If direction cannot be obtained from the data user, the laboratory must spike at least one sample in duplicate per extraction batch of up to 20 samples with the analytes in Table 1. Spiked sample results should be reported only to the data user whose sample was spiked, or as requested or required by a regulatory/control authority, or in a permit.
8.3.1. If, as in compliance monitoring, the concentration of a specific analyte will be checked against a regulatory concentration limit, the concentration of the spike should be at that limit; otherwise, the concentration of the spike should be one to five times higher than the background concentration determined in section 8.3.2, at or near the midpoint of the calibration range, or at the concentration in the LCS (section 8.4) whichever concentration would be larger. When no information is available, the mid-point of the calibration may be used.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of the each analyte of interest. If necessary to meet the requirement in section 8.3.1, prepare a new check sample concentrate (section 8.2.1) appropriate for the background concentration. Spike and analyze two additional sample aliquots of the same volume as the original sample, and determine the concentrations after spiking (A1 and A2) of each analyte. Calculate the percent recoveries (P1 and P2) as:
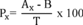 where T
is the known true value of the spike.
where T
is the known true value of the spike.
Also calculate the relative percent difference (RPD) between the concentrations (A1 and A2):
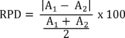
8.3.3 Compare the percent recoveries (P1 and P2) and the RPD for each analyte in the MS/MSD aliquots with the corresponding QC acceptance criteria for recovery (P) and RPD in Table 4.
(a) If any individual P falls outside the designated range for recovery in either aliquot, or the RPD limit is exceeded, the result for the analyte in the unspiked sample is suspect and may not be reported or used for permitting or regulatory compliance. See section 8.1.7 for disposition of failures.
(b) For analytes in Table 2 not listed in Table 4, QC acceptance criteria must be developed by the laboratory. EPA has provided guidance for development of QC acceptance criteria (References 12 and 13).
8.3.4 After analysis of a minimum of 20 MS/MSD samples for each target analyte and surrogate, and if the laboratory chooses to develop and apply optional in-house QC limits, the laboratory should calculate and apply the optional in-house QC limits for recovery and RPD of future MS/MSD samples (Section 8.3). The optional in-house QC limits for recovery are calculated as the mean observed recovery ±3 standard deviations, and the upper QC limit for RPD is calculated as the mean RPD plus 3 standard deviations of the RPDs. The in-house QC limits must be updated at least every two years and re-established after any major change in the analytical instrumentation or process. At least 80% of the analytes tested in the MS/MSD must have in-house QC acceptance criteria that are tighter than those in Table 4 and the remaining analytes (those not included in the 80%) must meet the acceptance criteria in Table 4. If an in-house QC limit for the RPD is greater than the limit in Table 4, then the limit in Table 4 must be used. Similarly, if an in-house lower limit for recovery is below the lower limit in Table 4, then the lower limit in Table 4 must be used, and if an in-house upper limit for recovery is above the upper limit in Table 4, then the upper limit in Table 4 must be used. The laboratory must evaluate surrogate recovery data in each sample against its in-house surrogate recovery limits. The laboratory may use 60 -140% as interim acceptance criteria for surrogate recoveries until in-house limits are developed. Alternatively, surrogate recovery limits may be developed from laboratory control charts. In-house QC acceptance criteria must be updated at least every two years.
8.4 Laboratory control sample (LCS) - A QC check sample (laboratory control sample, LCS; on-going precision and recovery sample, OPR) containing each single-component analyte of interest (section 1.4) must be extracted, concentrated, and analyzed with each extraction batch of up to 20 samples (section 3.1) to demonstrate acceptable recovery of the analytes of interest from a clean sample matrix. If multi-peak analytes are required, extract and prepare at least one as an LCS for each batch. Alternatively, the laboratory may set up a program where multi-peak LCS is rotated with a single-peak LCS.
8.4.1 Prepare the LCS by adding QC check sample concentrate (sections 6.8.3 and 8.2.1) to reagent water. Include all analytes of interest (section 1.4) in the LCS. The volume of reagent water must be the same as the nominal volume used for the sample, the DOC (Section 8.2), the blank (section 8.5), and the MS/MSD (section 8.3). Also add a volume of the surrogate solution (section 6.8.6).
8.4.2 Analyze the LCS prior to analysis of samples in the extraction batch (Section 3.1). Determine the concentration (A) of each analyte. Calculate the percent recovery as:
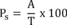 where T
is the true value of the concentration in the LCS.
where T
is the true value of the concentration in the LCS.
8.4.3 For each analyte, compare the percent recovery (P) with its corresponding QC acceptance criterion in Table 4. For analytes of interest in Table 2 not listed in Table 4, use the QC acceptance criteria developed for the MS/MSD (section 8.3.3.2), or limits based on laboratory control charts. If the recoveries for all analytes of interest fall within the designated ranges, analysis of blanks and field samples may proceed. If any individual recovery falls outside the range, proceed according to section 8.4.4.
Note:The large number of analytes in Tables 1 and 2 present a substantial probability that one or more will fail the acceptance criteria when all analytes are tested simultaneously. Because a re-test is allowed in event of failure (sections 8.1.7 and 8.4.4), it may be prudent to extract and analyze two LCSs together and evaluate results of the second analysis against the QC acceptance criteria only if an analyte fails the first test.
8.4.4 Repeat the test only for those analytes that failed to meet the acceptance criteria (P). If these analytes now pass, system performance is acceptable and analysis of blanks and samples may proceed. Repeated failure, however, will confirm a general problem with the measurement system. If this occurs, repeat the test using a fresh LCS (section 8.2.1) or an LCS prepared with a fresh QC check sample concentrate (section 8.2.1), or perform and document system repair. Subsequent to analysis of the LCS prepared with a fresh sample concentrate, or to system repair, repeat the LCS test (Section 8.4). If failure of the LCS indicates a systemic problem with samples in the batch, re-extract and re-analyze the samples in the batch. See Section 8.1.7 for disposition of repeated failures.
8.4.5 After analysis of 20 LCS samples, and if the laboratory chooses to develop and apply optional in-house QC limits, the laboratory should calculate and apply the optional in-house QC limits for recovery of future LCS samples (section 8.4). Limits for recovery in the LCS should be calculated as the mean recovery ±3 standard deviations. A minimum of 80% of the analytes tested for in the LCS must have QC acceptance criteria tighter than those in Table 4, and the remaining analytes (those not included in the 80%) must meet the acceptance criteria in Table 4. If an in-house lower limit for recovery is lower than the lower limit in Table 4, the lower limit in Table 4 must be used, and if an in-house upper limit for recovery is higher than the upper limit in Table 4, the upper limit in Table 4 must be used. Many of the analytes and surrogates do not contain acceptance criteria. The laboratory should use 60-140% as interim acceptance criteria for recoveries of spiked analytes and surrogates that do not have recovery limits specified in Table 4, and at least 80% of the surrogates must meet the 60-140% interim criteria until in-house LCS and surrogate limits are developed. Alternatively, acceptance criteria for analytes that do not have recovery limits in Table 4 may be based on laboratory control charts. In-house QC acceptance criteria must be updated at least every two years.
8.5 Blank - Extract and analyze a blank with each extraction batch (section 3.1) to demonstrate that the reagents and equipment used for preparation and analysis are free from contamination.
8.5.1 Prepare the blank from reagent water and spike it with the surrogates. The volume of reagent water must be the same as the volume used for samples, the DOC (section 8.2), the LCS (section 8.4), and the MS/MSD (section 8.3). Extract, concentrate, and analyze the blank using the same procedures and reagents used for the samples, LCS, and MS/MSD in the batch. Analyze the blank immediately after analysis of the LCS (section 8.4) and prior to analysis of the MS/MSD and samples to demonstrate freedom from contamination.
8.5.2 If any analyte of interest is found in the blank at a concentration greater than the MDL for the analyte, at a concentration greater than one-third the regulatory compliance limit, or at a concentration greater than one-tenth the concentration in a sample in the batch (section 3.1), whichever is greatest, analysis of samples must be halted and samples in the batch must be re-extracted and the extracts reanalyzed. Samples in a batch must be associated with an uncontaminated blank before the results for those samples may be reported or used for permitting or regulatory compliance purposes. If re-testing of blanks results in repeated failures, the laboratory should document the failures and report the problem and failures with the data.
8.6 Surrogate recovery - The laboratory must spike all samples with the surrogate standard spiking solution (section 6.8.6) per section 10.2.2 or 10.4.2, analyze the samples, and calculate the percent recovery of each surrogate. QC acceptance criteria for surrogates must be developed by the laboratory (section 8.4). If any recovery fails its criterion, attempt to find and correct the cause of the failure, and if sufficient volume is available, re-extract another aliquot of the affected sample; otherwise, see section 8.1.7 for disposition of repeated failures.
8.7 As part of the QC program for the laboratory, it is suggested but not required that method accuracy for wastewater samples be assessed and records maintained. After analysis of five or more spiked wastewater samples as in Section 8.3, calculate the average percent recovery (X) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent interval from X−2sp to X+2sp. For example, if X = 90% and sp = 10%, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each analyte on a regular basis to ensure process control (e.g., after each 5-10 new accuracy measurements). If desired, statements of accuracy for laboratory performance, independent of performance on samples, may be developed using LCSs.
8.8 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with another dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Collect samples as grab samples in glass bottles, or in refrigerated bottles using automatic sampling equipment. Collect 1-L of ambient waters, effluents, and other aqueous samples. If high concentrations of the analytes of interest are expected (e.g., for untreated effluents or in-process waters), collect a smaller volume (e.g., 250 mL), but not less than 100 mL, in addition to the 1-L sample. Follow conventional sampling practices, except do not pre-rinse the bottle with sample before collection. Automatic sampling equipment must be as free as possible of polyvinyl chloride or other tubing or other potential sources of contamination. If needed, collect additional sample(s) for the MS/MSD (section 8.3).
9.2 Ice or refrigerate the sample at ≤6 °C from the time of collection until extraction, but do not freeze. If aldrin is to be determined and residual chlorine is present, add 80 mg/L of sodium thiosulfate but do not add excess. Any method suitable for field use may be employed to test for residual chlorine (Reference 14). If sodium thiosulfate interferes in the determination of the analytes, an alternative preservative (e.g., ascorbic acid or sodium sulfite) may be used.
9.3 Extract all samples within seven days of collection and completely analyze within 40 days of extraction (Reference 1). If the sample will not be extracted within 72 hours of collection, adjust the sample pH to a range of 5.0-9.0 with sodium hydroxide solution or sulfuric acid. Record the volume of acid or base used.
10. Sample Extraction10.1 This section contains procedures for separatory funnel liquid-liquid extraction (SFLLE, section 10.2), continuous liquid-liquid extraction (CLLE, section 10.4), and disk-based solid-phase extraction (SPE, section 10.5). SFLLE is faster, but may not be as effective as CLLE for extracting polar analytes. SFLLE is labor intensive and may result in formation of emulsions that are difficult to break. CLLE is less labor intensive, avoids emulsion formation, but requires more time (18-24 hours), more hood space, and may require more solvent. SPE can be faster, unless the particulate load in an aqueous sample is so high that it slows the filtration process. If an alternative extraction scheme to those detailed in this method is used, all QC tests must be performed and all QC acceptance criteria must be met with that extraction scheme as an integral part of this method.
10.2 Separatory funnel liquid-liquid extraction (SFLLE).
10.2.1 The SFLLE procedure below assumes a sample volume of 1 L. When a different sample volume is extracted, adjust the volume of methylene chloride accordingly.
10.2.2 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into the separatory funnel. Pipet the surrogate standard spiking solution (section 6.8.6) into the separatory funnel. If the sample will be used for the LCS or MS or MSD, pipet the appropriate QC check sample concentrate (section 8.3 or 8.4) into the separatory funnel. Mix well. If the sample arrives in a larger sample bottle, 1 L may be measured in a graduated cylinder, then added to the separatory funnel.
Note:Instances in which the sample is collected in an oversized bottle should be reported by the laboratory to the data user. Of particular concern is that fact that this practice precludes rinsing the empty bottle with solvent as described below, which could leave hydrophobic pesticides on the wall of the bottle, and underestimate the actual sample concentrations.
10.2.3 Add 60 mL of methylene chloride to the sample bottle, seal, and shake for 30 seconds to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for two minutes with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 minutes. If an emulsion forms and the emulsion interface between the layers is more than one-third the volume of the solvent layer, employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, use of phase-separation paper, centrifugation, salting, freezing, or other physical methods. Collect the methylene chloride extract in a flask. If the emulsion cannot be broken (recovery of less than 80% of the methylene chloride, corrected for the water solubility of methylene chloride), transfer the sample, solvent, and emulsion into the extraction chamber of a continuous extractor and proceed as described in section 10.4.
10.2.4 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the flask. Perform a third extraction in the same manner. Proceed to macro-concentration (section 10.3.1).
10.2.5 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to an appropriately sized graduated cylinder. Record the sample volume to the nearest 5 mL. Sample volumes may also be determined by weighing the container before and after extraction or filling to the mark with water.
10.3 Concentration.
10.3.1 Macro concentration.
10.3.1.1 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator so long as the requirements of section 8.2 are met.
10.3.1.2 Pour the extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the flask and column with 20-30 mL of methylene chloride to complete the quantitative transfer.
10.3.1.3 If no cleanup is to be performed on the sample, add 500 µL (0.5 mL) of isooctane to the extract to act as a keeper during concentration.
10.3.1.4 Add one or two clean boiling chips and attach a three-ball Snyder column to the K-D evaporative flask. Pre-wet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60-65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15-20 minutes. At the proper rate of evaporation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL or other determined amount, remove the K-D apparatus from the water bath and allow it to drain and cool for at least 10 minutes.
10.3.1.5 If the extract is to be cleaned up by sulfur removal or acid back extraction, remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of methylene chloride. A 5-mL syringe is recommended for this operation. Adjust the final volume to 10 mL in methylene chloride and proceed to sulfur removal (section 11.5) or acid back extraction (section 11.6). If the extract is to cleaned up using one of the other cleanup procedures or is to be injected into the GC, proceed to Kuderna-Danish micro-concentration (section 10.3.2) or nitrogen evaporation and solvent exchange (section 10.3.3).
10.3.2 Kuderna-Danish micro concentration - Add another one or two clean boiling chips to the concentrator tube and attach a two-ball micro-Snyder column. Pre-wet the Snyder column by adding about 0.5 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60-65 °C) so that the concentrator tube is partially immersed in hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 5-10 minutes. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches approximately 1 mL or other required amount, remove the K-D apparatus from the water bath and allow it to drain and cool for at least 10 minutes. Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with approximately 0.2 mL of methylene chloride, and proceed to section 10.3.3 for nitrogen evaporation and solvent exchange.
10.3.3 Nitrogen evaporation and solvent exchange - Extracts to be subjected to solid-phase cleanup (SPE) are exchanged into 1.0 mL of the SPE elution solvent (section 6.7.2.2). Extracts to be subjected to Florisil® or alumina cleanups are exchanged into hexane. Extracts that have been cleaned up and are ready for analysis are exchanged into isooctane or hexane, to match the solvent used for the calibration standards.
10.3.3.1 Transfer the vial containing the sample extract to the nitrogen evaporation (blowdown) device (section 5.2.5.2). Lower the vial into a 50-55 °C water bath and begin concentrating. During the solvent evaporation process, do not allow the extract to become dry. Adjust the flow of nitrogen so that the surface of the solvent is just visibly disturbed. A large vortex in the solvent may cause analyte loss.
10.3.3.2 Solvent exchange.
10.3.3.2.1 When the volume of the liquid is approximately 500 µL, add 2 to 3 mL of the desired solvent (SPE elution solvent for SPE cleanup, hexane for Florisil or alumina, or isooctane for final injection into the GC) and continue concentrating to approximately 500 µL. Repeat the addition of solvent and concentrate once more.
10.3.3.3.2 Adjust the volume of an extract to be cleaned up by SPE, Florisil®, or alumina to 1.0 mL. Proceed to extract cleanup (section 11).
10.3.3.3 Extracts that have been cleaned up and are ready for analysis - Adjust the final extract volume to be consistent with the volume extracted and the sensitivity desired. The goal is for a full-volume sample (e.g., 1-L) to have a final extract volume of 10 mL, but other volumes may be used.
10.3.4 Transfer the concentrated extract to a vial with fluoropolymer-lined cap. Seal the vial and label with the sample number. Store in the dark at room temperature until ready for GC analysis. If GC analysis will not be performed on the same day, store the vial in the dark at ≤6 °C. Analyze the extract by GC per the procedure in section 12.
10.4 Continuous liquid/liquid extraction (CLLE).
10.4.1 Use CLLE when experience with a sample from a given source indicates an emulsion problem, or when an emulsion is encountered using SFLLE. CLLE may be used for all samples, if desired.
10.4.2 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Transfer the sample to the continuous extractor and, using a pipet, add surrogate standard spiking solution. If the sample will be used for the LCS, MS, or MSD, pipet the appropriate check sample concentrate (section 8.2.1 or 8.3.2) into the separatory funnel. Mix well. Add 60 mL of methylene chloride to the sample bottle, seal, and shake for 30 seconds to rinse the inner surface. Transfer the solvent to the extractor.
10.4.3 Repeat the sample bottle rinse with two additional 50-100 mL portions of methylene chloride and add the rinses to the extractor.
10.4.4 Add a suitable volume of methylene chloride to the distilling flask (generally 200-500 mL) and sufficient reagent water to ensure proper operation of the extractor, and extract the sample for 18-24 hours. A shorter or longer extraction time may be used if all QC acceptance criteria are met. Test and, if necessary, adjust the pH of the water to a range of 5.0-9.0 during the second or third hour of the extraction. After extraction, allow the apparatus to cool, then detach the distilling flask. Dry, concentrate, solvent exchange, and transfer the extract to a vial with fluoropolymer-lined cap, per Section 10.3.
10.4.5 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to an appropriately sized graduated cylinder. Record the sample volume to the nearest 5 mL. Sample volumes may also be determined by weighing the container before and after extraction or filling to the mark with water.
10.5 Solid-phase extraction of aqueous samples. The steps in this section address the extraction of aqueous field samples using disk-based solid-phase extraction (SPE) media, based on an ATP approved by EPA in 1995 (Reference 20). This application of SPE is distinct from that used in this method for the cleanup of sample extracts in section 11.2. Analysts must be careful not to confuse the equipment, supplies, or the procedural steps from these two different uses of SPE.
Note:Changes to the extraction conditions described below may be made by the laboratory under the allowance for method flexibility described in section 8.1, provided that the performance requirements in section 8.2 are met. However, changes in SPE materials, formats, and solvents must meet the requirements in section 8.1.2 and its subsections.
10.5.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. If the sample contains particulates, let stand to settle out the particulates before extraction.
10.5.2 Extract the sample as follows:
10.5.2.1 Place a 90-mm standard filter apparatus on a vacuum filtration flask or manifold and attach to a vacuum source. The vacuum gauge must read at least 25 in. of mercury when all valves are closed. Position a 90-mm C18 extraction disk onto the filter screen. Wet the entire disk with methanol. To aid in filtering samples with particulates, a 1-µm glass fiber filter or Empore® Filter Aid 400 can be placed on the top of the disk and wetted with methanol. Install the reservoir and clamp. Resume vacuum to dry the disk. Interrupt the vacuum. Wash the disk and reservoir with 20 mL of methylene chloride. Resume the vacuum briefly to pull methylene chloride through the disk. Interrupt the vacuum and allow the disk to soak for about a minute. Resume vacuum and completely dry the disk.
10.5.2.2 Condition the disk with 20 mL of methanol. Apply vacuum until nearly all the solvent has passed through the disk, interrupting it while solvent remains on the disk. Allow the disk to soak for about a minute. Resume vacuum to pull most of the methanol through, but interrupting it to leave a layer of methanol on the surface of the disk. Do not allow disk to dry. For uniform flow and good recovery, it is critical the disk not be allowed to dry from now until the end of the extraction. Discard waste solvent. Rinse the disk with 20 mL of deionized water. Resume vacuum to pull most of the water through, but interrupt it to leave a layer of water on the surface of the disk. Do not allow the disk to dry. If disk does dry, recondition with methanol as above.
10.5.2.3 Add the water sample to the reservoir and immediately apply the vacuum. If particulates have settled in the sample, gently decant the clear layer into the apparatus until most of the sample has been processed. Then pour the remainder including the particulates into the reservoir. Empty the sample bottle completely. When the filtration is complete, dry the disk for three minutes. Turn off the vacuum.
10.5.3 Discard sample filtrate. Insert tube to collect the eluant. The tube should fit around the drip tip of the base. Reassemble the apparatus. Add 5.0 mL of acetone to the center of the disk, allowing it to spread evenly over the disk. Turn the vacuum on and quickly off when the filter surface nears dryness but still remains wet. Allow to soak for 15 seconds. Add 20 mL of methylene chloride to the sample bottle, seal and shake to rinse the inside of the bottle. Transfer the methylene chloride from the bottle to the filter. Resume the vacuum slowly so as to avoid splashing.
Interrupt the vacuum when the filter surface nears dryness but still remains wet. Allow disk to soak in solvent for 20 seconds. Rinse the reservoir glass and disk with 10 mL of methylene chloride. Resume vacuum slowly. Interrupt vacuum when disk is covered with solvent. Allow to soak for 20 seconds. Resume vacuum to dry the disk. Remove the sample tube.
10.5.4 Dry, concentrate, solvent exchange, and transfer the extract to a vial with fluoropolymer-lined cap, per section 10.3.
10.5.5 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to an appropriately sized graduated cylinder. Record the sample volume to the nearest 5 mL. Sample volumes may also be determined by weighing the container before and after extraction or filling to the mark with water.
11. Extract Cleanup11.1 Cleanup may not be necessary for relatively clean samples (e.g., treated effluents, groundwater, drinking water). If particular circumstances require the use of a cleanup procedure, the laboratory may use any or all of the procedures below or any other appropriate procedure (e.g., gel permeation chromatography). However, the laboratory must first repeat the tests in sections 8.2, 8.3, and 8.4 to demonstrate that the requirements of those sections can be met using the cleanup procedure(s) as an integral part of this method. This is particularly important when the target analytes for the analysis include any of the single component pesticides in Table 2, because some cleanups have not been optimized for all of those analytes.
11.1.1 The solid-phase cartridge (section 11.2) removes polar organic compounds such as phenols.
11.1.2 The Florisil® column (section 11.3) allows for selected fractionation of the organochlorine analytes and will also eliminate polar interferences.
11.1.3 Alumina column cleanup (section 11.4) also removes polar materials.
11.1.4 Elemental sulfur, which interferes with the electron capture gas chromatography of some of the pesticides, may be removed using activated copper, or TBA sulfite. Sulfur removal (section 11.5) is required when sulfur is known or suspected to be present. Some chlorinated pesticides which also contain sulfur may be removed by this cleanup.
11.1.5 Acid back extraction (section 11.6) may be useful for cleanup of PCBs and other compounds not adversely affected by sulfuric acid.
11.2 Solid-phase extraction (SPE) as a cleanup. In order to use the C18 SPE cartridge in section 5.5.3.5 as a cleanup procedure, the sample extract must be exchanged from methylene chloride to methylene chloride:acetonitrile:hexane (50:3:47). Follow the solvent exchange steps in section 10.3.3.2 prior to attempting solid-phase cleanup.
Note:This application of SPE is distinct from that used in this method for the extraction of aqueous samples in section 10.5. Analysts must be careful not to confuse the equipment, supplies, or procedural steps from these two different uses of SPE.
11.2.1 Setup.
11.2.1.1 Attach the VacElute Manifold (section 5.5.3.2) to a water aspirator or vacuum pump with the trap and gauge installed between the manifold and vacuum source.
11.2.1.2 Place the SPE cartridges in the manifold, turn on the vacuum source, and adjust the vacuum to 5 to 10 psi.
11.2.2 Cartridge washing - Pre-elute each cartridge prior to use sequentially with 10-mL portions each of hexane, methanol, and water using vacuum for 30 seconds after each eluting solvent. Follow this pre-elution with 1 mL methylene chloride and three 10-mL portions of the elution solvent (section 6.7.2.2) using vacuum for 5 minutes after each eluting solvent. Tap the cartridge lightly while under vacuum to dry between solvent rinses. The three portions of elution solvent may be collected and used as a cartridge blank, if desired. Finally, elute the cartridge with 10 mL each of methanol and water, using the vacuum for 30 seconds after each eluant.
11.2.3 Extract cleanup.
11.2.3.1 After cartridge washing (section 11.2.2), release the vacuum and place the rack containing the 50-mL volumetric flasks (section 5.5.3.4) in the vacuum manifold. Re-establish the vacuum at 5 to 10 psi.
11.2.3.2 Using a pipette or a 1-mL syringe, transfer 1.0 mL of extract to the SPE cartridge. Apply vacuum for five minutes to dry the cartridge. Tap gently to aid in drying.
11.2.3.3 Elute each cartridge into its volumetric flask sequentially with three 10-mL portions of the methylene chloride:acetonitrile:hexane (50:3:47) elution solvent (section 6.7.2.2), using vacuum for five minutes after each portion. Collect the eluants in the 50-mL volumetric flasks.
11.2.3.4 Release the vacuum and remove the 50-mL volumetric flasks.
11.2.3.5 Concentrate the eluted extracts per Section 10.3.
11.3 Florisil®. In order to use Florisil cleanup, the sample extract must be exchanged from methylene chloride to hexane. Follow the solvent exchange steps in section 10.3.3.2 prior to attempting Florisil® cleanup.
Note:Alternative formats for this cleanup may be used by the laboratory, including cartridges containing Florisil®. If an alternative format is used, consult the manufacturer's instructions and develop a formal documented procedure to replace the steps in section 11.3 of this method and demonstrate that the alternative meets the relevant quality control requirements of this method.
11.3.1 If the chromatographic column does not contain a frit at the bottom, place a small plug of pre-cleaned glass wool in the column (section 5.2.4) to retain the Florisil®. Place the mass of Florisil® (nominally 20 g) predetermined by calibration (section 7.8 and Table 6) in a chromatographic column. Tap the column to settle the Florisil® and add 1 to 2 cm of granular anhydrous sodium sulfate to the top.
11.3.2 Add 60 mL of hexane to wet and rinse the sodium sulfate and Florisil®. Just prior to exposure of the sodium sulfate layer to the air, stop the elution of the hexane by closing the stopcock on the chromatographic column. Discard the eluant.
11.3.3 Transfer the concentrated extract (section 10.3.3) onto the column. Complete the transfer with two 1-mL hexane rinses, drawing the extract and rinses down to the level of the sodium sulfate.
11.3.4 Place a clean 500-mL K-D flask and concentrator tube under the column. Elute Fraction 1 with 200 mL of 6% (v/v) ethyl ether in hexane at a rate of approximately 5 mL/min. Remove the K-D flask and set it aside for later concentration. Elute Fraction 2 with 200 mL of 15% (v/v) ethyl ether in hexane into a second K-D flask. Elute Fraction 3 with 200 mL of 50% (v/v) ethyl ether in hexane into a third K-D flask. The elution patterns for the pesticides and PCBs are shown in Table 6.
11.3.5 Concentrate the fractions as in Section 10.3, except use hexane to prewet the column and set the water bath at about 85 °C. When the apparatus is cool, remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with hexane. Adjust the volume of Fraction 1 to approximately 10 mL for sulfur removal (Section 11.5), if required; otherwise, adjust the volume of the fractions to 10 mL, 1.0 mL, or other volume needed for the sensitivity desired. Analyze the concentrated extract by gas chromatography (Section 12).
11.4 Alumina. The sample extract must be exchanged from methylene chloride to hexane. Follow the solvent exchange steps in section 10.3.3.2 prior to attempting alumina cleanup.
11.4.1 If the chromatographic column does not contain a frit at the bottom, place a small plug of pre-cleaned glass wool in the chromatographic column (section 5.2.4) to retain the alumina. Add 10 g of alumina (section 6.7.3) on top of the plug. Tap the column to settle the alumina. Place 1-2 g of anhydrous sodium sulfate on top of the alumina.
11.4.2 Close the stopcock and fill the column to just above the sodium sulfate with hexane. Add 25 mL of hexane. Open the stopcock and adjust the flow rate of hexane to approximately 2 mL/min. Do not allow the column to go dry throughout the elutions.
11.4.3 When the level of the hexane is at the top of the column, quantitatively transfer the extract to the column. When the level of the extract is at the top of the column, slowly add 25 mL of hexane and elute the column to the level of the sodium sulfate. Discard the hexane.
11.4.4 Place a K-D flask (section 5.2.5.1.2) under the column and elute the pesticides with approximately 150 mL of hexane:ethyl ether (80:20 v/v). It may be necessary to adjust the volume of elution solvent for slightly different alumina activities.
11.4.5 Concentrate the extract per section 10.3.
11.5 Sulfur removal - Elemental sulfur will usually elute in Fraction 1 of the Florisil® column cleanup. If Florisil® cleanup is not used, or to remove sulfur from any of the Florisil® fractions, use one of the sulfur removal procedures below. These procedures may be applied to extracts in hexane, ethyl ether, or methylene chloride.
Note:Separate procedures using copper or TBA sulfite are provided in this section for sulfur removal. They may be used separately or in combination, if desired.
11.5.1 Removal with copper (Reference 15).
Note:Some of the analytes in Table 2 are not amenable to sulfur removal with copper (e.g., atrazine and diazinon). Therefore, before using copper to remove sulfur from an extract that will be analyzed for any of the non-PCB analytes in Table 2, the laboratory must demonstrate that the analytes can be extracted from an aqueous sample matrix that contains sulfur and recovered from an extract treated with copper. Acceptable performance can be demonstrated through the preparation and analysis of a matrix spike sample that meets the QC requirements for recovery.
11.5.1.1 Quantitatively transfer the extract to a 40- to 50-mL flask or bottle. If there is evidence of water in the K-D or round-bottom flask after the transfer, rinse the flask with small portions of hexane:acetone (40:60) and add to the flask or bottle. Mark and set aside the concentration flask for future use.
11.5.1.2 Add 10-20 g of granular anhydrous sodium sulfate to the flask. Swirl to dry the extract.
11.5.1.3 Add activated copper (section 6.7.4.1.4) and allow to stand for 30-60 minutes, swirling occasionally. If the copper does not remain bright, add more and swirl occasionally for another 30-60 minutes.
11.5.1.4 After drying and sulfur removal, quantitatively transfer the extract to a nitrogen-evaporation vial or tube and proceed to section 10.3.3 for nitrogen evaporation and solvent exchange, taking care to leave the sodium sulfate and copper foil in the flask.
11.5.2 Removal with TBA sulfite.
11.5.2.1 Using small volumes of hexane, quantitatively transfer the extract to a 40- to 50-mL centrifuge tube with fluoropolymer-lined screw cap.
11.5.2.2 Add 1-2 mL of TBA sulfite reagent (section 6.7.4.2.4), 2-3 mL of 2-propanol, and approximately 0.7 g of sodium sulfite (section 6.7.4.2.2) crystals to the tube. Cap and shake for 1-2 minutes. If the sample is colorless or if the initial color is unchanged, and if clear crystals (precipitated sodium sulfite) are observed, sufficient sodium sulfite is present. If the precipitated sodium sulfite disappears, add more crystalline sodium sulfite in approximately 0.5-g portions until a solid residue remains after repeated shaking.
11.5.2.3 Add 5-10 mL of reagent water and shake for 1-2 minutes. Centrifuge to settle the solids.
11.5.2.4 Quantitatively transfer the hexane (top) layer through a small funnel containing a few grams of granular anhydrous sodium sulfate to a nitrogen-evaporation vial or tube and proceed to section 10.3.3 for micro-concentration and solvent exchange.
11.6 Acid back extraction (section 6.1.2).
11.6.1 Quantitatively transfer the extract (section 10.3.1.5) to a 250-mL separatory funnel.
11.6.2 Partition the extract against 50 mL of sulfuric acid solution (section 6.1.2). Discard the aqueous layer. Repeat the acid washing until no color is visible in the aqueous layer, to a maximum of four washings.
11.6.3 Partition the extract against 50 mL of sodium chloride solution (section 6.7.5). Discard the aqueous layer.
11.6.4 Proceed to section 10.3.3 for micro-concentration and solvent exchange.
12. Gas Chromatography12.1 Establish the same operating conditions used in section 7.1 for instrument calibration.
12.2 If the internal standard calibration procedure is used, add the internal standard solution (section 6.9.3) to the extract as close as possible to the time of injection to minimize the possibility of loss by evaporation, adsorption, or reaction. For example, add 1 µL of 10 µg/mL internal standard solution into the extract, assuming no dilutions. Mix thoroughly.
12.3 Simultaneously inject an appropriate volume of the sample extract or standard solution onto both columns, using split, splitless, solvent purge, large-volume, or on-column injection. Alternatively, if using a single-column GC configuration, inject an appropriate volume of the sample extract or standard solution onto each GC column independently. If the sample is injected manually, the solvent-flush technique should be used. The injection volume depends upon the technique used and the sensitivity needed to meet MDLs or reporting limits for regulatory compliance. Injection volumes must be the same for all extracts. Record the volume injected to the nearest 0.05 µL.
12.4 Set the data system or GC control to start the temperature program upon sample injection, and begin data collection after the solvent peak elutes. Set the data system to stop data collection after the last analyte is expected to elute and to return the column to the initial temperature.
12.5 Perform all qualitative and quantitative measurements as described in Sections 14 and 15. When standards and extracts are not being used for analyses, store them refrigerated at <6 °C, protected from light, in screw-cap vials equipped with un-pierced fluoropolymer-lined septa.
13. System and Laboratory Performance13.1 At the beginning of each shift during which standards or extracts are analyzed, GC system performance and calibration must be verified for all analytes and surrogates on both column/detector systems. Adjustment and/or recalibration (per section 7) are performed until all performance criteria are met. Only after all performance criteria are met may samples, blanks and other QC samples, and standards be analyzed.
13.2 Inject an aliquot of the calibration verification standard (section 6.8.4) on both columns. Inject an aliquot of each of the multi-component standards.
13.3 Retention times - The absolute retention times of the peak maxima shall be within ±2 seconds of the retention times in the calibration verification (section 7.8).
13.4 GC resolution - Resolution is acceptable if the valley height between two peaks (as measured from the baseline) is less than 40% of the shorter of the two peaks.
13.4.1 DB-608 column - DDT and endrin aldehyde
13.4.2 DB-1701 column - alpha and gamma chlordane
Note:If using other GC columns or stationary phases, these resolution criteria apply to these four target analytes and any other closely eluting analytes on those other GC columns.
13.5 Decomposition of DDT and endrin - If DDT, endrin, or their breakdown products are to be determined, this test must be performed prior to calibration verification (section 13.6). DDT decomposes to DDE and DDD. Endrin decomposes to endrin aldehyde and endrin ketone.
13.5.1 Inject 1 µL of the DDT and endrin decomposition solution (section 6.8.7). As noted in section 6.8.7, other injection volumes may be used as long as the concentrations of DDT and endrin in the solution are adjusted to introduce the masses of the two analytes into the instrument that are listed in section 6.8.7.
13.5.2 Measure the areas of the peaks for DDT, DDE, DDD, endrin, endrin aldehyde, and endrin ketone in the chromatogram and calculate the percent breakdown as shown in the equations below:

13.5.3 Both the % breakdown of DDT and of endrin must be less than 20%, otherwise the system is not performing acceptably for DDT and endrin. In this case, repair the GC column system that failed and repeat the performance tests (sections 13.2 to 13.6) until the specification is met.
Note:DDT and endrin decomposition are usually caused by accumulations of particulates in the injector and in the front end of the column. Cleaning and silanizing the injection port liner, and breaking off a short section of the front end of the column will usually eliminate the decomposition problem. Either of these corrective actions may affect retention times, GC resolution, and calibration linearity.
13.6 Calibration verification.
13.6.1 Compute the percent recovery of each analyte and of the coeluting analytes, based on the initial calibration data (section 7.5 or 7.6).
13.6.2 For each analyte or for coeluting analytes, compare the concentration with the limits for calibration verification in Table 4. For coeluting analytes, use the coeluting analyte with the least restrictive specification (the widest range). For analytes in Table 2 not listed in Table 4, QC acceptance criteria must be developed by the laboratory. EPA has provided guidance for development of QC acceptance criteria (References 13 and 14). If the recoveries for all analytes meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may continue. If, however, any recovery falls outside the calibration verification range, system performance is unacceptable for that analyte. If this occurs, repair the system and repeat the test (section 13.6), or prepare a fresh calibration standard and repeat the test, or recalibrate (section 7). See Section 8.1.7 for information on repeated test failures.
13.7 Laboratory control sample.
13.7.1 Analyze the extract of the LCS (section 6.8.3) extracted with each sample batch (Section 8.4). See Section 8.4 for criteria acceptance of the LCS.
13.7.2 It is suggested, but not required, that the laboratory update statements of data quality. Add results that pass the specifications in section 13.7.3 to initial (section 8.7) and previous ongoing data. Update QC charts to form a graphic representation of continued laboratory performance. Develop a statement of laboratory data quality for each analyte by calculating the average percent recovery (R) and the standard deviation of percent recovery, sr. Express the accuracy as a recovery interval from R − 2sr to R + 2sr. For example, if R = 95% and sr = 5%, the accuracy is 85 to 105%.
13.8 Internal standard response - If internal standard calibration is used, verify that detector sensitivity has not changed by comparing the response (area or height) of each internal standard in the sample, blank, LCS, MS, and MSD to the response in calibration verification (section 6.8.3). The peak area or height of the internal standard should be within 50% to 200% ( 1/2 to 2x) of its respective peak area or height in the verification standard. If the area or height is not within this range, compute the concentration of the analytes using the external standard method (section 7.5). If the analytes are affected, re-prepare and reanalyze the sample, blank, LCS, MS, or MSD, and repeat the pertinent test.
14. Qualitative Identification14.1 Identification is accomplished by comparison of data from analysis of a sample, blank, or other QC sample with data from calibration verification (section 7.7.1 or 13.5), and with data stored in the retention-time and calibration libraries (section 7.7). The retention time window is determined as described in section 14.2. Identification is confirmed when retention time agrees on both GC columns, as described below. Alternatively, GC/MS identification may be used to provide another means of identification.
14.2 Establishing retention time windows.
14.2.1 Using the data from the multi-point initial calibration (section 7.4), determine the retention time in decimal minutes (not minutes:seconds) of each peak representing a single-component target analyte on each column/detector system. For the multi-component analytes, use the retention times of the five largest peaks in the chromatograms on each column/detector system.
14.2.2 Calculate the standard deviation of the retention times for each single-component analyte on each column/detector system and for the three to five exclusive (unique large) peaks for each multi-component analyte.
14.2.3 Define the width of the retention time window as three times that standard deviation. Establish the center of the retention time window for each analyte by using the absolute retention time for each analyte from the calibration verification standard at the beginning of the analytical shift. For samples run during the same shift as an initial calibration, use the retention time of the mid-point standard of the initial calibration. If the calculated RT window is less than 0.02 minutes, then use 0.02 minutes as the window.
Note:Procedures for establishing retention time windows from other sources may be employed provided that they are clearly documented and provide acceptable performance. Such performance may be evaluated using the results for the spiked QC samples described in this method, such as laboratory control samples and matrix spike samples.
14.2.4 The retention time windows must be recentered when a new GC column is installed or if a GC column has been shortened during maintenance to a degree that the retention times of analytes in the calibration verification standard have shifted close to the lower limits of the established retention time windows.
14.2.5 RT windows should be checked periodically by examining the peaks in spiked samples such as the LCS or MS/MSD to confirm that peaks for known analytes are properly identified.
14.2.6 If the retention time of an analyte in the calibration (Section 7.4) varies by more than 5 seconds across the calibration range as a function of the concentration of the standard, using the standard deviation of the retention times (section 14.2.3) to set the width of the retention time window may not adequately serve to identify the analyte in question under routine conditions. In such cases, data from additional analyses of standards may be required to adequately model the chromatographic behavior of the analyte.
14.3 Identifying the analyte in a sample.
14.3.1 In order to identify a single-component analyte from analysis of a sample, blank, or other QC sample, the peak representing the analyte must fall within its respective retention time windows on both column/detector systems (as defined in section 14.2). That identification is further supported by the comparison of the numerical results on both columns, as described in section 15.7.
14.3.2 In order to identify a multi-component analyte, pattern matching (fingerprinting) may be used, or the three to five exclusive (unique and largest) peaks for that analyte must fall within their respective retention time windows on both column/detector systems (as defined in section 14.2). That identification is further supported by the comparison of the numerical results on both columns, as described in section 15.7. Alternatively, GC/MS identification may be used. Differentiation among some of the Aroclors may require evaluation of more than five peaks to ensure correct identification.
14.4 GC/MS confirmation. When the concentration of an analyte is sufficient and the presence or identity is suspect, its presence should be confirmed by GC/MS. In order to match the sensitivity of the GC/ECD, confirmation would need to be by GC/MS-SIM, or the estimated concentration would need to be 100 times higher than the GC/ECD calibration range. The extract may be concentrated by an additional amount to allow a further attempt at GC/MS confirmation.
14.5 Additional information that may aid the laboratory in the identification of an analyte. The occurrence of peaks eluting near the retention time of an analyte of interest increases the probability of a false positive for the analyte. If the concentration is insufficient for confirmation by GC/MS, the laboratory may use the cleanup procedures in this method (section 11) on a new sample aliquot to attempt to remove the interferent. After attempts at cleanup are exhausted, the following steps may be helpful to assure that the substance that appears in the RT windows on both columns is the analyte of interest.
14.5.1 Determine the consistency of the RT data for the analyte on each column. For example, if the RT is very stable (i.e., varies by no more than a few seconds) for the calibration, calibration verification, blank, LCS, and MS/MSD, the RT for the analyte of interest in the sample should be within this variation regardless of the window established in Section 14.2. If the analyte is not within this variation on both columns, it is likely not present.
14.5.2 The possibility exists that the RT for the analyte in a sample could shift if extraneous materials are present. This possibility may be able to be confirmed or refuted by the behavior of the surrogates in the sample. If multiple surrogates are used that span the length of the chromatographic run, the RTs for the surrogates on both columns are consistent with their RTs in calibration, calibration verification, blank, LCS, and MS/MSD, it is unlikely that the RT for the analyte of interest has shifted.
14.5.3 If the RT for the analyte is shifted slightly later on one column and earlier on the other, and the surrogates have not shifted, it is highly unlikely that the analyte is present, because shifts nearly always occur in the same direction on both columns.
15. Quantitative Determination15.1 External standard quantitation - Calculate the concentration of the analyte in the extract using the calibration curve or average calibration factor determined in calibration (section 7.5.2) and the following equation:
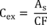 where:
Cex = Concentration of the analyte in the extract (ng/mL) As = Peak
height or area for the analyte in the standard or sample CF =
Calibration factor, as defined in Section 7.5.1
where:
Cex = Concentration of the analyte in the extract (ng/mL) As = Peak
height or area for the analyte in the standard or sample CF =
Calibration factor, as defined in Section 7.5.1
15.2 Internal standard quantitation - Calculate the concentration of the analyte in the extract using the calibration curve or average response factor determined in calibration (section 7.6.2) and the following equation:
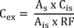 where:
Cex = Concentration of the analyte in the extract (ng/mL) As = Peak
height or area for the analyte in the standard or sample Cis =
Concentration of the internal standard (ng/mL) Ais = Area of the
internal standard RF = Response factor, as defined in section 7.6.1
where:
Cex = Concentration of the analyte in the extract (ng/mL) As = Peak
height or area for the analyte in the standard or sample Cis =
Concentration of the internal standard (ng/mL) Ais = Area of the
internal standard RF = Response factor, as defined in section 7.6.1
15.3 Calculate the concentration of the analyte in the sample using the concentration in the extract, the extract volume, the sample volume, and the dilution factor, per the following equation:
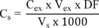 where: Cs
= Concentration of the analyte in the sample (µg/L) Vex = Final
extract volume (mL) Cex = Concentration in the extract (ng/mL) Vs =
Volume of sample (L) DF = Dilution factor and the factor of 1,000
in the denominator converts the final units from ng/L to µg/L
where: Cs
= Concentration of the analyte in the sample (µg/L) Vex = Final
extract volume (mL) Cex = Concentration in the extract (ng/mL) Vs =
Volume of sample (L) DF = Dilution factor and the factor of 1,000
in the denominator converts the final units from ng/L to µg/L
15.4 If the concentration of any target analyte exceeds the calibration range, either extract and analyze a smaller sample volume, or dilute and analyze the diluted extract.
15.5 Quantitation of multi-component analytes.
15.5.1 PCBs as Aroclors. Quantify an Aroclor by comparing the sample chromatogram to that of the most similar Aroclor standard as indicated in section 14.3.2. Compare the responses of 3 to 5 major peaks in the calibration standard for that Aroclor with the peaks observed in the sample extract. The amount of Aroclor is calculated using the individual calibration factor for each of the 3 to 5 characteristic peaks chosen in section 7.5.1. Determine the concentration of each of the characteristic peaks, using the average calibration factor calculated for that peak in section 7.5.2, and then those 3 to 5 concentrations are averaged to determine the concentration of that Aroclor.
15.5.2 Other multi-component analytes. Quantify any other multi-component analytes (technical chlordane or toxaphene) using the same peaks used to develop the average calibration factors in section 7.5.2. Determine the concentration of each of the characteristic peaks, and then the concentrations represented by those characteristic peaks are averaged to determine the concentration of the analyte. Alternatively, for toxaphene, the analyst may determine the calibration factor in section 7.5.2 by summing the areas of all of the peaks for the analyte and using the summed of the peak areas in the sample chromatogram to determine the concentration. However, the approach used for toxaphene must be the same for the calibration and the sample analyses.
15.6 Reporting of results. As noted in section 1.6.1, EPA has promulgated this method at 40 CFR part 136 for use in wastewater compliance monitoring under the National Pollutant Discharge Elimination System (NPDES). The data reporting practices described here are focused on such monitoring needs and may not be relevant to other uses of the method.
15.6.1 Report results for wastewater samples in µg/L without correction for recovery. (Other units may be used if required by in a permit.) Report all QC data with the sample results.
15.6.2 Reporting level. Unless specified otherwise by a regulatory authority or in a discharge permit, results for analytes that meet the identification criteria are reported down to the concentration of the ML established by the laboratory through calibration of the instrument (see section 7.5 or 7.6 and the glossary for the derivation of the ML). EPA considers the terms “reporting limit,” “quantitation limit,” and “minimum level” to be synonymous.
15.6.2.1 Report the lower result from the two columns (see section 15.7 below) for each analyte in each sample or QC standard at or above the ML to 3 significant figures. Report a result for each analyte in each sample or QC standard below the ML as “<ML,” where “ML” is the concentration of the analyte at the ML (e.g., if the ML is 10 µg/L, then report the result as <10 µg/L), or as required by the regulatory authority or permit. Report a result for each analyte in a blank at or above the MDL to 2 significant figures. Report a result for each analyte found in a blank below the MDL as “<MDL,” where MDL is the concentration of the analyte at the MDL, or as required by the regulatory/control authority or permit.
15.6.2.2 In addition to reporting results for samples and blank(s) separately, the concentration of each analyte in a blank or field blank associated with that sample may be subtracted from the result for that sample, but only if requested or required by a regulatory authority or in a permit. In this case, both the sample result and the blank results must be reported together.
15.6.2.3 Report the result for an analyte in a sample or extract that has been diluted at the least dilute level at which the peak area is within the calibration range (i.e., above the ML for the analyte) and the MS/MSD recovery and RPD are within their respective QC acceptance criteria (Table 4). This may require reporting results for some analytes from different analyses. Results for each analyte in MS/MSD samples should be reported from the same GC column as used to report the results for that analyte in the unspiked sample. If the MS/MSD recoveries and RPDs calculated in this manner do not meet the acceptance criteria in Table 4, the analyst may use the results from the other GC column to determine if the MS/MSD results meet the acceptance criteria. If such a situation occurs, the results for the sample should be recalculated using the same GC column data as used for the MS/MSD samples, and reported with appropriate annotations that alert the data user of the issue.
15.6.2.4 Results from tests performed with an analytical system that is not in control (i.e., that does not meet acceptance criteria for all of QC tests in this method) must not be reported or otherwise used for permitting or regulatory compliance purposes, but do not relieve a discharger or permittee of reporting timely results. See section 8.1.7 for dispositions of failures. If the holding time would be exceeded for a re-analysis of the sample, the regulatory/control authority should be consulted for disposition.
15.6.3 Analyze the sample by GC/MS or on a third column when analytes have co-eluted or interfere with determination on both columns.
Note:Dichlone and kepone do not elute from the DB-1701 column and must be confirmed on a DB-5 column, or by GC/MS.
15.7 Quantitative information that may aid in the confirmation of the presence of an analyte.
15.7.1 As noted in Section 14.3, the relative agreement between the numerical results from the two GC columns may be used to support the identification of the target analyte by providing evidence that co-eluting interferences are not present at the retention time of the target analyte. Calculate the percent difference (%D) between the results for the analyte from both columns, as follows:
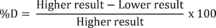
In general, if the %D of the two results is less than 50% (e.g., a factor of 2), then the pesticide is present. This %D is generous and allows for the pesticide that has the largest measurement error.
Note:Laboratories may employ metrics less than 50% for this comparison, including those specified in other analytical methods for these pesticides (e.g., CLP or SW-846).
15.7.2 If the amounts do not agree, and the RT data indicate the presence of the analyte (per Section 14), it is likely that a positive interference is present on the column that yielded the higher result. That interferent may be represented by a separate peak on the other column that does not coincide with the retention time of any of the target analytes. If the interfering peak is evident on the other column, report the result from that column and advise the data user that the interference resulted in a %D value greater than 50%. If an interferent is not identifiable on the second column, then the results must be reported as “not detected” at the lower concentration. In this event, the pesticide is not confirmed and the reporting limit is elevated. See section 8.1.7 for disposition of problem results.
Note:The resulting elevation of the reporting limit may not meet the requirements for compliance monitoring and the use of additional cleanup procedures may be required.
16. Analysis of Complex Samples16.1 Some samples may contain high levels (greater than 1 µg/L) of the analytes of interest, interfering analytes, and/or polymeric materials. Some samples may not concentrate to 1.0 mL (section 10.3.3.3.2); others may overload the GC column and/or detector.
16.2 When an interference is known or suspected to be present, the laboratory should attempt to clean up the sample extract using the SPE cartridge (section 11.2), by Florisil® (Section 11.3), Alumina (Section 11.4), sulfur removal (section 11.5), or another clean up procedure appropriate to the analytes of interest. If these techniques do not remove the interference, the extract is diluted by a known factor and reanalyzed (section 12). Dilution until the extract is lightly colored is preferable. Typical dilution factors are 2, 5, and 10.
16.3 Recovery of surrogate(s) - In most samples, surrogate recoveries will be similar to those from reagent water. If surrogate recovery is outside the limits developed in Section 8.6, re-extract and reanalyze the sample if there is sufficient sample and if it is within the 7-day extraction holding time. If surrogate recovery is still outside this range, extract and analyze one-tenth the volume of sample to overcome any matrix interference problems. If a sample is highly colored or suspected to be high in concentration, a 1-L sample aliquot and a 100-mL sample aliquot could be extracted simultaneously and still meet the holding time criteria, while providing information about a complex matrix.
16.4 Recovery of the matrix spike and matrix spike duplicate (MS/MSD) - In most samples, MS/MSD recoveries will be similar to those from reagent water. If either the MS or MSD recovery is outside the range specified in Section 8.3.3, one-tenth the volume of sample is spiked and analyzed. If the matrix spike recovery is still outside the range, the result for the unspiked sample may not be reported or used for permitting or regulatory compliance purposes. See Section 8.1.7 for dispositions of failures. Poor matrix spike recovery does not relieve a discharger or permittee of reporting timely results.
17. Method Performance17.1 This method was tested for linearity of spike recovery from reagent water and has been demonstrated to be applicable over the concentration range from 4x MDL to 1000x MDL with the following exceptions: Chlordane recovery at 4x MDL was low (60%); Toxaphene recovery was demonstrated linear over the range of 10x MDL to 1000x MDL (Reference 3).
17.2 The 1984 version of this method was tested by 20 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations (Reference 2). Concentrations used in the study ranged from 0.5 to 30 µg/L for single-component pesticides and from 8.5 to 400 µg/L for multi-component analytes. These data are for a subset of analytes described in the current version of the method.
17.3 During the development of Method 1656, a similar EPA procedure for the organochlorine pesticides, single-operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the analyte and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 5.
18. Pollution Prevention18.1 Pollution prevention encompasses any technique that reduces or eliminates the quantity or toxicity of waste at the point of generation. Many opportunities for pollution prevention exist in laboratory operations. EPA has established a preferred hierarchy of environmental management techniques that places pollution prevention as the management option of first choice. Whenever feasible, the laboratory should use pollution prevention techniques to address waste generation. When wastes cannot be reduced at the source, the Agency recommends recycling as the next best option.
18.2 The analytes in this method are used in extremely small amounts and pose little threat to the environment when managed properly. Standards should be prepared in volumes consistent with laboratory use to minimize the disposal of excess volumes of expired standards. This method utilizes significant quantities of methylene chloride. Laboratories are encouraged to recover and recycle this and other solvents during extract concentration.
18.3 For information about pollution prevention that may be applied to laboratories and research institutions, consult “Less is Better: Laboratory Chemical Management for Waste Reduction” (Reference 19), available from the American Chemical Society's Department of Governmental Relations and Science Policy, 1155 16th Street NW., Washington DC 20036, 202-872-4477.
19. Waste Management19.1 The laboratory is responsible for complying with all Federal, State, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions, and to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance is also required with any sewage discharge permits and regulations. An overview of requirements can be found in Environmental Management Guide for Small Laboratories (EPA 233-B-98-001).
19.2 Samples at pH <2, or pH >12, are hazardous and must be handled and disposed of as hazardous waste, or neutralized and disposed of in accordance with all federal, state, and local regulations. It is the laboratory's responsibility to comply with all federal, state, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions. The laboratory using this method has the responsibility to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance is also required with any sewage discharge permits and regulations. For further information on waste management, see “The Waste Management Manual for Laboratory Personnel,” also available from the American Chemical Society at the address in section 18.3.
19.3 Many analytes in this method decompose above 500 °C. Low-level waste such as absorbent paper, tissues, animal remains, and plastic gloves may be burned in an appropriate incinerator. Gross quantities of neat or highly concentrated solutions of toxic or hazardous chemicals should be packaged securely and disposed of through commercial or governmental channels that are capable of handling toxic wastes.
19.4 For further information on waste management, consult The Waste Management Manual for Laboratory Personnel and Less is Better-Laboratory Chemical Management for Waste Reduction, available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street NW., Washington, DC 20036, 202-872-4477.
20. References 1. “Determination of Pesticides and PCBs in Industrial and Municipal Wastewaters,” EPA 600/4-82-023, National Technical Information Service, PB82-214222, Springfield, Virginia 22161, April 1982. 2. “EPA Method Study 18 Method 608-Organochlorine Pesticides and PCBs,” EPA 600/4-84-061, National Technical Information Service, PB84-211358, Springfield, Virginia 22161, June 1984. 3. “Method Detection Limit and Analytical Curve Studies, EPA Methods 606, 607, and 608,” Special letter report for EPA Contract 68-03-2606, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, June 1980. 4. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practice for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia. 5. Giam, C.S., Chan, H.S., and Nef, G.S. “Sensitive Method for Determination of Phthalate Ester Plasticizers in Open-Ocean Biota Samples,” Analytical Chemistry, 47:2225 (1975). 6. Giam, C.S. and Chan, H.S. “Control of Blanks in the Analysis of Phthalates in Air and Ocean Biota Samples,” U.S. National Bureau of Standards, Special Publication 442, pp. 701-708, 1976. 7. Solutions to Analytical Chemistry Problems with Clean Water Act Methods, EPA 821-R-07-002, March 2007. 8. “Carcinogens-Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977. 9. “Occupational Exposure to Hazardous Chemicals in Laboratories,” (29 CFR 1910.1450), Occupational Safety and Health Administration, OSHA. 10. 40 CFR 136.6(b)(4)(j). 11. Mills, P.A. “Variation of Florisil Activity: Simple Method for Measuring Absorbent Capacity and Its Use in Standardizing Florisil Columns,” Journal of the Association of Official Analytical Chemists, 51:29, (1968). 12. 40 CFR 136.6(b)(2)(i). 13. Protocol for EPA Approval of New Methods for Organic and Inorganic Analytes in Wastewater and Drinking Water (EPA-821-B-98-003) March 1999. 14. Methods 4500 Cl F and 4500 Cl G, Standard Methods for the Examination of Water and Wastewater, published jointly by the American Public Health Association, American Water Works Association, and Water Environment Federation, 1015 Fifteenth St., Washington, DC 20005, 20th Edition, 2000. 15. “Manual of Analytical Methods for the Analysis of Pesticides in Human and Environmental Samples,” EPA-600/8-80-038, U.S. Environmental Protection Agency, Health Effects Research Laboratory, Research Triangle Park, North Carolina. 16. USEPA, 2000, Method 1656 Organo-Halide Pesticides In Wastewater, Soil, Sludge, Sediment, and Tissue by GC/HSD, EPA-821-R-00-017, September 2000. 17. USEPA, 2010, Method 1668C Chlorinated Biphenyl Congeners in Water, Soil, Sediment, Biosolids, and Tissue by HRGC/HRMS, EPA-820-R-10-005, April 2010. 18. USEPA, 2007, Method 1699: Pesticides in Water, Soil, Sediment, Biosolids, and Tissue by HRGC/HRMS, EPA-821-R-08-001, December 2007. 19. “Less is Better,” American Chemical Society on-line publication, http://www.acs.org/content/dam/acsorg/about/governance/committees/chemicalsafety/publications/less-is-better.pdf. 20. EPA Method 608 ATP 3M0222, An alternative test procedure for the measurement of organochlorine pesticides and polychlorinated biphenyls in waste water. Federal Register, Vol. 60, No. 148 August 2, 1995. 21. TablesTable 1 - Pesticides 1
| Analyte | CAS No. | MDL 2 (ng/L) |
ML 3 (ng/L) |
|---|---|---|---|
| Aldrin | 309-00-2 | 4 | 12 |
| alpha-BHC | 319-84-6 | 3 | 9 |
| beta-BHC | 319-85-7 | 6 | 18 |
| delta-BHC | 319-86-8 | 9 | 27 |
| gamma-BHC (Lindane) | 58-89-9 | 4 | 12 |
| alpha-Chlordane 4 | 5103-71-9 | 14 | 42 |
| gamma-Chlordane 4 | 5103-74-2 | 14 | 42 |
| 4,4′-DDD | 72-54-8 | 11 | 33 |
| 4,4′-DDE | 72-55-9 | 4 | 12 |
| 4,4′-DDT | 50-29-3 | 12 | 36 |
| Dieldrin | 60-57-1 | 2 | 6 |
| Endosulfan I | 959-98-8 | 14 | 42 |
| Endosulfan II | 33213-65-9 | 4 | 12 |
| Endosulfan sulfate | 1031-07-8 | 66 | 198 |
| Endrin | 72-20-8 | 6 | 18 |
| Endrin aldehyde | 7421-93-4 | 23 | 70 |
| Heptachlor | 76-44-8 | 3 | 9 |
| Heptachlor epoxide | 1024-57-3 | 83 | 249 |
1 All analytes in this table are Priority Pollutants (40 CFR part 423, appendix A).
2 40 CFR part 136, appendix B, June 30, 1986.
3 ML = Minimum Level - see Glossary for definition and derivation, calculated as 3 times the MDL.
4 MDL based on the MDL for Chlordane.
Table 2 - Additional Analytes
| Analyte | CAS No. | MDL 3 (ng/L) |
ML 4 (ng/L) |
|---|---|---|---|
| Acephate | 30560-19-1 | ||
| Alachlor | 15972-60-8 | ||
| Atrazine | 1912-24-9 | ||
| Benfluralin (Benefin) | 1861-40-1 | ||
| Bromacil | 314-40-9 | ||
| Bromoxynil octanoate | 1689-99-2 | ||
| Butachlor | 23184-66-9 | ||
| Captafol | 2425-06-1 | ||
| Captan | 133-06-2 | ||
| Carbophenothion (Trithion) | 786-19-6 | ||
| Chlorobenzilate | 510-15-6 | ||
| Chloroneb (Terraneb) | 2675-77-6 | ||
| Chloropropylate (Acaralate) | 5836-10-2 | ||
| Chlorothalonil | 1897-45-6 | ||
| Cyanazine | 21725-46-2 | ||
| DCPA (Dacthal) | 1861-32-1 | ||
| 2,4′-DDD | 53-19-0 | ||
| 2,4′-DDE | 3424-82-6 | ||
| 2,4′-DDT | 789-02-6 | ||
| Diallate (Avadex) | 2303-16-4 | ||
| 1,2-Dibromo-3-chloropropane (DBCP) | 96-12-8 | ||
| Dichlone | 117-80-6 | ||
| Dichloran | 99-30-9 | ||
| Dicofol | 115-32-2 | ||
| Endrin ketone | 53494-70-5 | ||
| Ethalfluralin (Sonalan) | 55283-68-6 | ||
| Etridiazole | 2593-15-9 | ||
| Fenarimol (Rubigan) | 60168-88-9 | ||
| Hexachlorobenzene 1 | 118-74-1 | ||
| Hexachlorocyclopentadiene 1 | 77-47-4 | ||
| Isodrin | 465-73-6 | ||
| Isopropalin (Paarlan) | 33820-53-0 | ||
| Kepone | 143-50-0 | ||
| Methoxychlor | 72-43-5 | ||
| Metolachlor | 51218-45-2 | ||
| Metribuzin | 21087-64-9 | ||
| Mirex | 2385-85-5 | ||
| Nitrofen (TOK) | 1836-75-5 | ||
| cis-Nonachlor | 5103-73-1 | ||
| trans-Nonachlor | 39765-80-5 | ||
| Norfluorazon | 27314-13-2 | ||
| Octachlorostyrene | 29082-74-4 | ||
| Oxychlordane | 27304-13-8 | ||
| PCNB (Pentachloronitrobenzene) | 82-68-8 | ||
| Pendamethalin (Prowl) | 40487-42-1 | ||
| cis-Permethrin | 61949-76-6 | ||
| trans-Permethrin | 61949-77-7 | ||
| Perthane (Ethylan) | 72-56-0 | ||
| Propachlor | 1918-16-7 | ||
| Propanil | 709-98-8 | ||
| Propazine | 139-40-2 | ||
| Quintozene | 82-68-8 | ||
| Simazine | 122-34-9 | ||
| Strobane | 8001-50-1 | ||
| Technazene | 117-18-0 | ||
| Technical Chlordane 2 | |||
| Terbacil | 5902-51-2 | ||
| Terbuthylazine | 5915-41-3 | ||
| Toxaphene 1 | 8001-35-2 | 240 | 720 |
| Trifluralin | 1582-09-8 | ||
| PCB-1016 1 | 12674-11-2 | ||
| PCB-1221 1 | 11104-28-2 | ||
| PCB-1232 1 | 11141-16-5 | ||
| PCB-1242 1 | 53469-21-9 | 65 | 95 |
| PCB-1248 1 | 12672-29-6 | ||
| PCB-1254 1 | 11097-69-1 | ||
| PCB-1260 1 | 11096-82-5 | ||
| PCB-1268 | 11100-14-4 |
1 Priority Pollutants (40 CFR part 423, appendix A).
2 Technical Chlordane may be used in cases where historical reporting has only been for this form of Chlordane.
3 40 CFR part 136, appendix B, June 30, 1986.
4 ML = Minimum Level - see Glossary for definition and derivation, calculated as 3 times the MDL.
Table 3 - Example Retention Times 1
| Analyte | Retention
time (min) 2 |
|
|---|---|---|
| DB-608 | DB-1701 | |
| Acephate | 5.03 | ( 3) |
| Trifluralin | 5.16 | 6.79 |
| Ethalfluralin | 5.28 | 6.49 |
| Benfluralin | 5.53 | 6.87 |
| Diallate-A | 7.15 | 6.23 |
| Diallate-B | 7.42 | 6.77 |
| alpha-BHC | 8.14 | 7.44 |
| PCNB | 9.03 | 7.58 |
| Simazine | 9.06 | 9.29 |
| Atrazine | 9.12 | 9.12 |
| Terbuthylazine | 9.17 | 9.46 |
| gamma-BHC (Lindane) | 9.52 | 9.91 |
| beta-BHC | 9.86 | 11.90 |
| Heptachlor | 10.66 | 10.55 |
| Chlorothalonil | 10.66 | 10.96 |
| Dichlone | 10.80 | ( 4) |
| Terbacil | 11.11 | 12.63 |
| delta-BHC | 11.20 | 12.98 |
| Alachlor | 11.57 | 11.06 |
| Propanil | 11.60 | 14.10 |
| Aldrin | 11.84 | 11.46 |
| DCPA | 12.18 | 12.09 |
| Metribuzin | 12.80 | 11.68 |
| Triadimefon | 12.99 | 13.57 |
| Isopropalin | 13.06 | 13.37 |
| Isodrin | 13.47 | 11.12 |
| Heptachlor epoxide | 13.97 | 12.56 |
| Pendamethalin | 14.21 | 13.46 |
| Bromacil | 14.39 | ( 3) |
| alpha-Chlordane | 14.63 | 14.20 |
| Butachlor | 15.03 | 15.69 |
| gamma-Chlordane | 15.24 | 14.36 |
| Endosulfan I | 15.25 | 13.87 |
| 4,4′-DDE | 16.34 | 14.84 |
| Dieldrin | 16.41 | 15.25 |
| Captan | 16.83 | 15.43 |
| Chlorobenzilate | 17.58 | 17.28 |
| Endrin | 17.80 | 15.86 |
| Nitrofen (TOK) | 17.86 | 17.47 |
| Kepone | 17.92 | ( 3 5) |
| 4,4′-DDD | 18.43 | 17.77 |
| Endosulfan II | 18.45 | 18.57 |
| Bromoxynil octanoate | 18.85 | 18.57 |
| 4,4′-DDT | 19.48 | 18.32 |
| Carbophenothion | 19.65 | 18.21 |
| Endrin aldehyde | 19.72 | 19.18 |
| Endosulfan sulfate | 20.21 | 20.37 |
| Captafol | 22.51 | 21.22 |
| Norfluorazon | 20.68 | 22.01 |
| Mirex | 22.75 | 19.79 |
| Methoxychlor | 22.80 | 20.68 |
| Endrin ketone | 23.00 | 21.79 |
| Fenarimol | 24.53 | 23.79 |
| cis-Permethrin | 25.00 | 23.59 |
| trans-Permethrin | 25.62 | 23.92 |
| PCB-1016 | ||
| PCB-1221 | ||
| PCB-1232 | ||
| PCB-1242 | ||
| PCB-1248 | ||
| PCB-1254 | ||
| PCB-1260 (5 peaks) | 15.44 | 14.64 |
| 15.73 | 15.36 | |
| 16.94 | 16.53 | |
| 17.28 | 18.70 | |
| 19.17 | 19.92 | |
| Toxaphene (5 peaks) | 16.60 | 16.60 |
| 17.37 | 17.52 | |
| 18.11 | 17.92 | |
| 19.46 | 18.73 | |
| 19.69 | 19.00 | |
1 Data from EPA Method 1656 (Reference 16).
2 Columns: 30-m long x 0.53-mm ID fused-silica capillary; DB-608, 0.83 µm; and DB-1701, 1.0 µm.
Conditions suggested to meet retention times shown: 150 °C for 0.5 minute, 150-270 °C at 5 °C/min, and 270 °C until trans-Permethrin elutes.
Carrier gas flow rates approximately 7 mL/min.
3 Does not elute from DB-1701 column at level tested.
4 Not recovered from water at the levels tested.
5 Dichlone and Kepone do not elute from the DB-1701 column and should be confirmed on DB-5.
Table 4 - QC Acceptance Criteria
| Analyte | Calibration verification (%) |
Test concentration (µg/L) |
Limit for s (% SD) |
Range for X (%) |
Range for P (%) |
Maximum MS/MSD RPD (%) |
|---|---|---|---|---|---|---|
| Aldrin | 75-125 | 2.0 | 25 | 54-130 | 42-140 | 35 |
| alpha-BHC | 69-125 | 2.0 | 28 | 49-130 | 37-140 | 36 |
| beta-BHC | 75-125 | 2.0 | 38 | 39-130 | 17-147 | 44 |
| delta-BHC | 75-125 | 2.0 | 43 | 51-130 | 19-140 | 52 |
| gamma-BHC | 75-125 | 2.0 | 29 | 43-130 | 32-140 | 39 |
| alpha-Chlordane | 73-125 | 50.0 | 24 | 55-130 | 45-140 | 35 |
| gamma-Chlordane | 75-125 | 50.0 | 24 | 55-130 | 45-140 | 35 |
| 4,4′-DDD | 75-125 | 10.0 | 32 | 48-130 | 31-141 | 39 |
| 4,4′-DDE | 75-125 | 2.0 | 30 | 54-130 | 30-145 | 35 |
| 4,4′-DDT | 75-125 | 10.0 | 39 | 46-137 | 25-160 | 42 |
| Dieldrin | 48-125 | 2.0 | 42 | 58-130 | 36-146 | 49 |
| Endosulfan I | 75-125 | 2.0 | 25 | 57-141 | 45-153 | 28 |
| Endosulfan II | 75-125 | 10.0 | 63 | 22-171 | D-202 | 53 |
| Endosulfan sulfate | 70-125 | 10.0 | 32 | 38-132 | 26-144 | 38 |
| Endrin | 5-125 | 10.0 | 42 | 51-130 | 30-147 | 48 |
| Heptachlor | 75-125 | 2.0 | 28 | 43-130 | 34-140 | 43 |
| Heptachlor epoxide | 75-125 | 2.0 | 22 | 57-132 | 37-142 | 26 |
| Toxaphene | 68-134 | 50.0 | 30 | 56-130 | 41-140 | 41 |
| PCB-1016 | 75-125 | 50.0 | 24 | 61-103 | 50-140 | 36 |
| PCB-1221 | 75-125 | 50.0 | 50 | 44-150 | 15-178 | 48 |
| PCB-1232 | 75-125 | 50.0 | 32 | 28-197 | 10-215 | 25 |
| PCB-1242 | 75-125 | 50.0 | 26 | 50-139 | 39-150 | 29 |
| PCB-1248 | 75-125 | 50.0 | 32 | 58-140 | 38-158 | 35 |
| PCB-1254 | 75-125 | 50.0 | 34 | 44-130 | 29-140 | 45 |
| PCB-1260 | 75-125 | 50.0 | 28 | 37-130 | 8-140 | 38 |
S = Standard deviation of four recovery measurements for the DOC (section 8.2.4).
X = Average of four recovery measurements for the DOC (section 8.2.4).
P = Recovery for the LCS (section 8.4.3).
Note: These criteria were developed from data in Table 5 (Reference 2). Where necessary, limits for recovery have been broadened to assure applicability to concentrations below those in Table 5.
Table 5 - Precision and Recovery as Functions of Concentration
| Analyte | Recovery, X′ (µg/L) |
Single analyst precision, sr′ (µg/L) |
Overall precision, S′ (µg/L) |
|---|---|---|---|
| Aldrin | 0.81C + 0.04 | 0.16(X) − 0.04 | 0.20(X) − 0.01 |
| alpha-BHC | 0.84C + 0.03 | 0.13(X) + 0.04 | 0.23(X) − 0.00 |
| beta-BHC | 0.81C + 0.07 | 0.22(X) − 0.02 | 0.33(X) − 0.05 |
| delta-BHC | 0.81C + 0.07 | 0.18(X) + 0.09 | 0.25(X) + 0.03 |
| gamma-BHC (Lindane) | 0.82C − 0.05 | 0.12(X) + 0.06 | 0.22(X) + 0.04 |
| Chlordane | 0.82C − 0.04 | 0.13(X) + 0.13 | 0.18(X) + 0.18 |
| 4,4′-DDD | 0.84C + 0.30 | 0.20(X) − 0.18 | 0.27(X) − 0.14 |
| 4,4′-DDE | 0.85C + 0.14 | 0.13(X) + 0.06 | 0.28(X) − 0.09 |
| 4,4′-DDT | 0.93C − 0.13 | 0.17(X) + 0.39 | 0.31(X) − 0.21 |
| Dieldrin | 0.90C + 0.02 | 0.12(X) + 0.19 | 0.16(X) + 0.16 |
| Endosulfan I | 0.97C + 0.04 | 0.10(X) + 0.07 | 0.18(X) + 0.08 |
| Endosulfan II | 0.93C + 0.34 | 0.41(X) − 0.65 | 0.47(X) − 0.20 |
| Endosulfan sulfate | 0.89C − 0.37 | 0.13(X) + 0.33 | 0.24(X) + 0.35 |
| Endrin | 0.89C − 0.04 | 0.20(X) + 0.25 | 0.24(X) + 0.25 |
| Heptachlor | 0.69C + 0.04 | 0.06(X) + 0.13 | 0.16(X) + 0.08 |
| Heptachlor epoxide | 0.89C + 0.10 | 0.18(X) − 0.11 | 0.25(X) − 0.08 |
| Toxaphene | 0.80C + 1.74 | 0.09(X) + 3.20 | 0.20(X) + 0.22 |
| PCB-1016 | 0.81C + 0.50 | 0.13(X) + 0.15 | 0.15(X) + 0.45 |
| PCB-1221 | 0.96C + 0.65 | 0.29(X) − 0.76 | 0.35(X) − 0.62 |
| PCB-1232 | 0.91C + 10.8 | 0.21(X) − 1.93 | 0.31(X) + 3.50 |
| PCB-1242 | 0.93C + 0.70 | 0.11(X) + 1.40 | 0.21(X) + 1.52 |
| PCB-1248 | 0.97C + 1.06 | 0.17(X) + 0.41 | 0.25(X) − 0.37 |
| PCB-1254 | 0.76C + 2.07 | 0.15(X) + 1.66 | 0.17(X) + 3.62 |
| PCB-1260 | 0.66C + 3.76 | 0.22(X) − 2.37 | 0.39(X) − 4.86 |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
Table 6 - Distribution of Chlorinated Pesticides and PCBs Into Florisil® Column Fractions
| Analyte | Percent Recovery by Fraction 1 | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| Aldrin | 100 | ||
| alpha-BHC | 100 | ||
| beta-BHC | 97 | ||
| delta-BHC | 98 | ||
| gamma-BHC (Lindane) | 100 | ||
| Chlordane | 100 | ||
| 4,4′-DDD | 99 | ||
| 4,4′-DDE | 98 | ||
| 4,4′-DDT | 100 | ||
| Dieldrin | 0 | 100 | |
| Endosulfan I | 37 | 64 | |
| Endosulfan II | 0 | 7 | 91 |
| Endosulfan sulfate | 0 | 0 | 106 |
| Endrin | 4 | 96 | |
| Endrin aldehyde | 0 | 68 | 26 |
| Heptachlor | 100 | ||
| Heptachlor epoxide | 100 | ||
| Toxaphene | 96 | ||
| PCB-1016 | 97 | ||
| PCB-1221 | 97 | ||
| PCB-1232 | 95 | 4 | |
| PCB-1242 | 97 | ||
| PCB-1248 | 103 | ||
| PCB-1254 | 90 | ||
| PCB-1260 | |||
1 Eluant composition:
Fraction 1 - 6% ethyl ether in hexane.
Fraction 2 - 15% ethyl ether in hexane.
Fraction 3 - 50% ethyl ether in hexane.
Table 7 - Suggested Calibration Groups 1
| Analyte |
|---|
| Calibration Group 1: |
| Acephate |
| Alachlor |
| Atrazine |
| beta-BHC |
| Bromoxynil octanoate |
| Captafol |
| Diallate |
| Endosulfan sulfate |
| Endrin |
| Isodrin |
| Pendimethalin (Prowl) |
| trans-Permethrin |
| Calibration Group 2: |
| alpha-BHC |
| DCPA |
| 4,4′-DDE |
| 4,4′-DDT |
| Dichlone |
| Ethalfluralin |
| Fenarimol |
| Methoxychlor |
| Metribuzin |
| Calibration Group 3: |
| gamma-BHC (Lindane) |
| gamma-Chlordane |
| Endrin ketone |
| Heptachlor epoxide |
| Isopropalin |
| Nitrofen (TOK) |
| PCNB |
| cis-Permethrin |
| Trifluralin |
| Callibration Group 4: |
| Benfluralin |
| Chlorobenzilate |
| Dieldrin |
| Endosulfan I |
| Mirex |
| Terbacil |
| Terbuthylazine |
| Triadimefon |
| Calibration Group 5: |
| alpha-Chlordane |
| Captan |
| Chlorothalonil |
| 4,4′-DDD |
| Norfluorazon |
| Simazine |
| Calibration Group 6: |
| Aldrin |
| delta-BHC |
| Bromacil |
| Butachlor |
| Endosulfan II |
| Heptachlor |
| Kepone |
| Calibration Group 7: |
| Carbophenothion |
| Chloroneb |
| Chloropropylate |
| DBCP |
| Dicofol |
| Endrin aldehyde |
| Etridiazone |
| Perthane |
| Propachlor |
| Propanil |
| Propazine |
1 The analytes may be organized in other calibration groups, provided that there are no coelution problems and that all QC requirements are met.
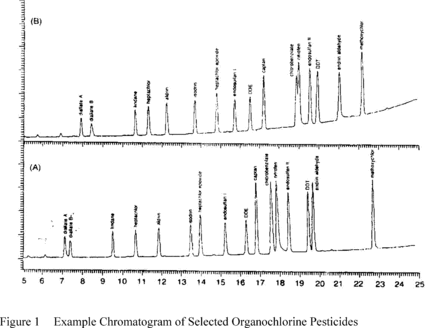
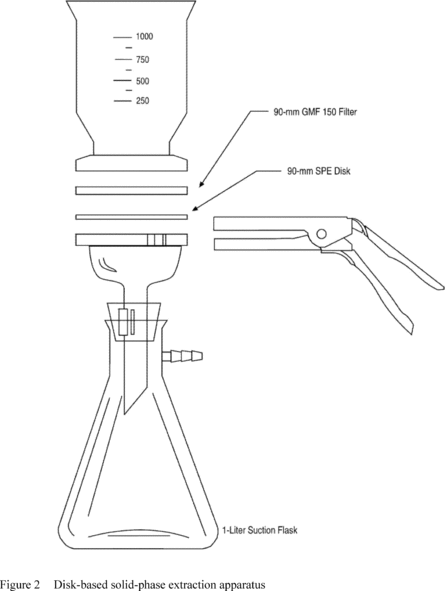 23. Glossary
23. Glossary
These definitions and purposes are specific to this method but have been conformed to common usage to the extent possible.
23.1 Units of weight and measure and their abbreviations.
23.1.1 Symbols.
°C degrees Celsius µg microgram µL microliter < less than ≤ less than or equal to > greater than % percent23.1.2 Abbreviations (in alphabetical order).
cm centimeter g gram hr hour ID inside diameter in. inch L liter M molar solution - one mole or gram molecular weight of solute in one liter of solution mg milligram min minute mL milliliter mm millimeter N Normality - one equivalent of solute in one liter of solution ng nanogram psia pounds-per-square inch absolute psig pounds-per-square inch gauge v/v volume per unit volume w/v weight per unit volume23.2 Definitions and acronyms (in alphabetical order)
Analyte - A compound or mixture of compounds (e.g., PCBs) tested for by this method. The analytes are listed in Tables 1 and 2.
Analytical batch - The set of samples analyzed on a given instrument during a 24-hour period that begins and ends with calibration verification (sections 7.8 and 13). See also “Extraction batch.”
Blank (method blank; laboratory blank) - An aliquot of reagent water that is treated exactly as a sample including exposure to all glassware, equipment, solvents, reagents, internal standards, and surrogates that are used with samples. The blank is used to determine if analytes or interferences are present in the laboratory environment, the reagents, or the apparatus.
Calibration factor (CF) - See section 7.5.1.
Calibration standard - A solution prepared from stock solutions and/or a secondary standards and containing the analytes of interest, surrogates, and internal standards. This standard is used to model the response of the GC instrument against analyte concentration.
Calibration verification - The process of confirming that the response of the analytical system remains within specified limits of the calibration.
Calibration verification standard - The standard (section 6.8.4) used to verify calibration (sections 7.8 and 13.6).
Extraction Batch - A set of up to 20 field samples (not including QC samples) started through the extraction process in a given 24-hour shift. Each extraction batch of 20 or fewer samples must be accompanied by a blank (section 8.5), a laboratory control sample (LCS, section 8.4), a matrix spike and duplicate (MS/MSD; section 8.3), resulting in a minimum of five samples (1 field sample, 1 blank, 1 LCS, 1 MS, and 1 MSD) and a maximum of 24 samples (20 field samples, 1 blank, 1 LCS, 1 MS, and 1 MSD) for the batch. If greater than 20 samples are to be extracted in a 24-hour shift, the samples must be separated into extraction batches of 20 or fewer samples.
Field Duplicates - Two samples collected at the same time and place under identical conditions, and treated identically throughout field and laboratory procedures. Results of analyses the field duplicates provide an estimate of the precision associated with sample collection, preservation, and storage, as well as with laboratory procedures.
Field blank - An aliquot of reagent water or other reference matrix that is placed in a sample container in the field, and treated as a sample in all respects, including exposure to sampling site conditions, storage, preservation, and all analytical procedures. The purpose of the field blank is to determine if the field or sample transporting procedures and environments have contaminated the sample. See also “Blank.”
GC - Gas chromatograph or gas chromatography.
Gel-permeation chromatography (GPC) - A form of liquid chromatography in which the analytes are separated based on exclusion from the solid phase by size.
Internal standard - A compound added to an extract or standard solution in a known amount and used as a reference for quantitation of the analytes of interest and surrogates. Also see Internal standard quantitation.
Internal standard quantitation - A means of determining the concentration of an analyte of interest (Tables 1 and 2) by reference to a compound not expected to be found in a sample.
IDC - Initial Demonstration of Capability (section 8.2); four aliquots of a reference matrix spiked with the analytes of interest and analyzed to establish the ability of the laboratory to generate acceptable precision and recovery. An IDC is performed prior to the first time this method is used and any time the method or instrumentation is modified.
Laboratory Control Sample (LCS; laboratory fortified blank; section 8.4) - An aliquot of reagent water spiked with known quantities of the analytes of interest and surrogates. The LCS is analyzed exactly like a sample. Its purpose is to assure that the results produced by the laboratory remain within the limits specified in this method for precision and recovery.
Laboratory Fortified Sample Matrix - See Matrix spike.
Laboratory reagent blank - See blank.
Matrix spike (MS) and matrix spike duplicate (MSD) (laboratory fortified sample matrix and duplicate) - Two aliquots of an environmental sample to which known quantities of the analytes of interest and surrogates are added in the laboratory. The MS/MSD are prepared and analyzed exactly like a field sample. Their purpose is to quantify any additional bias and imprecision caused by the sample matrix. The background concentrations of the analytes in the sample matrix must be determined in a separate aliquot and the measured values in the MS/MSD corrected for background concentrations.
May - This action, activity, or procedural step is neither required nor prohibited.
May not - This action, activity, or procedural step is prohibited.
Method detection limit (MDL) - A detection limit determined by the procedure at 40 CFR part 136, appendix B. The MDLs determined by EPA are listed in Tables 1 and 2. As noted in section 1.6, use the MDLs in Tables 1 and 2 in conjunction with current MDL data from the laboratory actually analyzing samples to assess the sensitivity of this procedure relative to project objectives and regulatory requirements (where applicable).
Minimum level (ML) - The term “minimum level” refers to either the sample concentration equivalent to the lowest calibration point in a method or a multiple of the method detection limit (MDL), whichever is higher. Minimum levels may be obtained in several ways: They may be published in a method; they may be based on the lowest acceptable calibration point used by a laboratory; or they may be calculated by multiplying the MDL in a method, or the MDL determined by a laboratory, by a factor of 3. For the purposes of NPDES compliance monitoring, EPA considers the following terms to be synonymous: “quantitation limit,” “reporting limit,” and “minimum level.”
MS - Mass spectrometer or mass spectrometry.
Must - This action, activity, or procedural step is required.
Preparation blank - See blank.
Reagent water - Water demonstrated to be free from the analytes of interest and potentially interfering substances at the MDLs for the analytes in this method.
Regulatory compliance limit - A limit on the concentration or amount of a pollutant or contaminant specified in a nationwide standard, in a permit, or otherwise established by a regulatory/control authority.
Relative standard deviation (RSD) - The standard deviation times 100 divided by the mean. Also termed “coefficient of variation.”
RF - Response factor. See section 7.6.2.
RPD - Relative percent difference.
RSD - See relative standard deviation.
Safety Data Sheet (SDS) - Written information on a chemical's toxicity, health hazards, physical properties, fire, and reactivity, including storage, spill, and handling precautions that meet the requirements of OSHA, 29 CFR 1910.1200(g) and appendix D to § 1910.1200. United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS), third revised edition, United Nations, 2009.
Should - This action, activity, or procedural step is suggested but not required.
SPE - Solid-phase extraction; a sample extraction or extract cleanup technique in which an analyte is selectively removed from a sample or extract by passage over or through a material capable of reversibly adsorbing the analyte.
Stock solution - A solution containing an analyte that is prepared using a reference material traceable to EPA, the National Institute of Science and Technology (NIST), or a source that will attest to the purity and authenticity of the reference material.
Surrogate - A compound unlikely to be found in a sample, which is spiked into the sample in a known amount before extraction, and which is quantified with the same procedures used to quantify other sample components. The purpose of the surrogate is to monitor method performance with each sample.
Method 609 - Nitroaromatics and Isophorone 1. Scope and Application1.1 This method covers the determination of certain nitroaromatics and isophorone. The following parameters may be determined by this method:
| Parameter | STORET No. | CAS No. |
|---|---|---|
| 2,4-Dinitrotoluene | 34611 | 121-14-2 |
| 2,6-Dinitrotoluene | 34626 | 606-20-2 |
| Isophorone | 34408 | 78-59-1 |
| Nitrobenzene | 34447 | 98-95-3 |
1.2 This is a gas chromatographic (GC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compounds above, compound identifications should be supported by at least one additional qualitative technique. This method describes analytical conditions for a second gas chromatographic column that can be used to confirm measurements made with the primary column. Method 625 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for all of the parameters listed above, using the extract produced by this method.
1.3 The method detection limit (MDL, defined in Section 14.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.4 The sample extraction and concentration steps in this method are essentially the same as in Methods 606, 608, 611, and 612. Thus, a single sample may be extracted to measure the parameters included in the scope of each of these methods. When cleanup is required, the concentration levels must be high enough to permit selecting aliquots, as necessary, to apply appropriate cleanup procedures. The analyst is allowed the latitude, under Section 12, to select chromatographic conditions appropriate for the simultaneous measurement of combinations of these parameters.
1.5 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.6 This method is restricted to use by or under the supervision of analysts experienced in the use of a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is extracted with methylene chloride using a separatory funnel. The methylene chloride extract is dried and exchanged to hexane during concentration to a volume of 10 mL or less. Isophorone and nitrobenzene are measured by flame ionization detector gas chromatography (FIDGC). The dinitrotoluenes are measured by electron capture detector gas chromatography (ECDGC). 2
2.2 The method provides a Florisil column cleanup procedure to aid in the elimination of interferences that may be encountered.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardware that lead to discrete artifacts and/or elevated baseliles in gas chromatograms. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 3 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Some thermally stable materials, such as PCBs, may not be eliminated by this treatment. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Thorough rinsing with such solvents usually eliminates PCB interference. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to minimize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Matrix interferences may be caused by contaminants that are co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. The cleanup procedure in Section 11 can be used to overcome many of these interferences, but unique samples may require additional cleanup approaches to achieve the MDL listed in Table 1.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 4-6 for the information of the analyst.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1-L or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flow meter is required to collect flow proportional composites.
5.2 Glassware (All specifications are suggested. Catalog numbers are included for illustration only.):
5.2.1 Separatory funnel - 2-L, with Teflon stopcock.
5.2.2 Drying column - Chromatographic column, approximately 400 mm long × 19 mm ID, with coarse frit filter disc.
5.2.3 Chromatographic column - 100 mm long × 10 mm ID, with Teflon stopcock.
5.2.4 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes K-570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. Ground glass stopper is used to prevent evaporation of extracts.
5.2.5 Evaporative flask, Kuderna-Danish - 500-mL (Kontes K-570001-0500 or equivalent). Attach to concentrator tube with springs.
5.2.6 Snyder column, Kuderna-Danish - Three-ball macro (Kontes K-503000-0121 or equivalent).
5.2.7 Snyder column, Kuderna-Danish - Two-ball micro (Kontes K-569001-0219 or equivalent).
5.2.8 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.3 Boiling chips - Approximately 10/40 mesh. Heat to 400 °C for 30 min or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 Balance - Analytical, capable of accurately weighing 0.0001 g.
5.6 Gas chromatograph - An analytical system complete with gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.6.1 Column 1 - 1.2 m long × 2 or 4 mm ID glass, packed with 1.95% QF-1/1.5% OV-17 on Gas-Chrom Q (80/100 mesh) or equivalent. This column was used to develop the method performance statements given in Section 14. Guidelines for the use of alternate column packings are provided in Section 12.1.
5.6.2 Column 2 - 3.0 m long × 2 or 4 mm ID glass, packed with 3% OV-101 on Gas-Chrom Q (80/100 mesh) or equivalent.
5.6.3 Detectors - Flame ionization and electron capture detectors. The flame ionization detector (FID) is used when determining isophorone and nitrobenzene. The electron capture detector (ECD) is used when determining the dinitrotoluenes. Both detectors have proven effective in the analysis of wastewaters and were used in develop the method performance statements in Section 14. Guidelines for the use to alternate detectors are provided in Section 12.1.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.2 Sodium hydroxide solution (10 N) - Dissolve 40 g of NaOH (ACS) in reagent water and dilute to 100 mL.
6.3 Sulfuric acid (1 + 1) - Slowly, add 50 mL of H2SO4 (ACS, sp. gr. 1.84) to 50 mL of reagent water.
6.4 Acetone, hexane, methanol, methylene chloride - Pesticide quality or equivalent.
6.5 Sodium sulfate - (ACS) Granular, anhydrous. Purify by heating at 400 °C for 4 h in a shallow tray.
6.6 Florisil - PR grade (60/100 mesh). Purchase activated at 1250 °F and store in dark in glass containers with ground glass stoppers or foil-lined screw caps. Before use, activate each batch at least 16 h at 200 °C in a foil-covered glass container and allow to cool.
6.7 Stock standard solutions (1.00 µg/µL) - Stock standard solutions can be prepared from pure standard materials or purchased as certified solutions.
6.7.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in hexane and dilute to volume in a 10-mL volumetric flask. Larger volumes can be used at the convenience of the analyst. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.7.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store at 4 °C and protect from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.7.3 Stock standard solutions must be replaced after six months, or sooner if comparison with check standards indicates a problem.
6.8 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Establish gas chromatographic operating conditions equivalent to those given in Table 1. The gas chromatographic system can be calibrated using the external standard technique (Section 7.2) or the internal standard technique (Section 7.3).
7.2 External standard calibration procedure:
7.2.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with hexane. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.2.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against the mass injected. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to amount injected (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD) linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.3 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flash. To each calibration standard, add a known constant amount of one or more internal standards, and dilute to volume with hexane. One of the standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.3.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against concentration for each compound and internal standard. Calculate response factors (RF) for each compound using Equation 1.
Equation 1.
where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of the parameter to be measured (µg/L).If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.
7.4 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of one or more calibration standards. If the response for any parameter varies from the predicted response by more than ±15%, a new calibration curve must be prepared for that compound.
7.5 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.4, 11.1, and 12.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Before processing any samples, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed, a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1,5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest in acetone at a concentration of 20 µg/mL for each dinitrotoluene and 100 µg/mL for isophorone and nitrobenzene. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at the test concentrations shown in Table 2 by adding 1.00 mL of QC check sample concentrate to each of four 1-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter. Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at the test concentration in Section 8.2.2 or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determile background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any; or, if none (2) the larger of either 5 times higher than the expected background concentration or the test concentration in Section 8.2.2.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100 (A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 7 If spiking was performed at a concentration lower than the test concentration in Section 8.2.2, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X 8; (3) calculate the range for recovery at the spike concentration as (100 X′/T) ±2.44 (100 S′/T)%. 7
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4. If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 1.0 mL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 1 L of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 2. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 8 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C from the time of collection until extraction.
9.3 All samples must be extracted within 7 days of collection and completely analyzed within 40 days of extraction. 2
10. Sample Extraction10.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel. Check the pH of the sample with wide-range pH paper and adjust to within the range of 5 to 9 with sodium hydroxide solution or sulfuric acid.
10.2 Add 60 mL of methylene chloride to the sample bottle, seal, and shake 30 s to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min. with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the methylene chloride extract in a 250-mL Erlenmeyer flask.
10.3 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.4 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator if the requirements of Section 8.2 are met.
10.5 Pour the combined extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the Erlenmeyer flask and column with 20 to 30 mL of methylene chloride to complete the quantitative transfer.
10.6 Sections 10.7 and 10.8 describe a procedure for exchanging the methylene chloride solvent to hexane while concentrating the extract volume to 1.0 mL. When it is not necessary to achieve the MDL in Table 2, the solvent exchange may be made by the addition of 50 mL of hexane and concentration to 10 mL as described in Method 606, Sections 10.7 and 10.8.
10.7 Add one or two clean boiling chips to the evaporative flask and attach a three-ball Snyder column. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
10.8 Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of methylene chloride. A 5-mL syringe is recommended for this operation. Add 1 to 2 mL of hexane and a clean boiling chip to the concentrator tube and attach a two-ball micro-Snyder column. Prewet the column by adding about 0.5 mL of hexane to the top. Place the micro-K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 5 to 10 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood. When the apparent volume of liquid reaches 0.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
10.9 Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with a minimum amount of hexane. Adjust the extract volume to 1.0 mL. Stopper the concentrator tube and store refrigerated if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial. If the sample extract requires no further cleanup, proceed with gas chromatographic analysis (Section 12). If the sample requires further cleanup, proceed to Section 11.
10.10 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1000-mL graduated cylinder. Record the sample volume to the nearest 5 mL.
11. Cleanup and Separation11.1 Cleanup procedures may not be necessary for a relatively clean sample matrix. If particular circumstances demand the use of a cleanup procedure, the analyst may use the procedure below or any other appropriate procedure. However, the analyst first must demonstrate that the requirements of Section 8.2 can be met using the method as revised to incorporate the cleanup procedure.
11.2 Florisil column cleanup:
11.2.1 Prepare a slurry of 10 g of activated Florisil in methylene chloride/hexane (1 + 9)(V/V) and place the Florisil into a chromatographic column. Tap the column to settle the Florisil and add 1 cm of anhydrous sodium sulfate to the top. Adjust the elution rate to about 2 mL/min.
11.2.2 Just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the sample extract onto the column using an additional 2 mL of hexane to complete the transfer. Just prior to exposure of the sodium sulfate layer to the air, add 30 mL of methylene chloride/hexane (1 + 9)(V/V) and continue the elution of the column. Discard the eluate.
11.2.3 Next, elute the column with 30 mL of acetone/methylene chloride (1 + 9)(V/V) into a 500-mL K-D flask equipped with a 10-mL concentrator tube. Concentrate the collected fraction as in Sections 10.6, 10.7, 10.8, and 10.9 including the solvent exchange to 1 mL of hexane. This fraction should contain the nitroaromatics and isophorone. Analyze by gas chromatography (Section 12).
12. Gas Chromatography12.1 Isophorone and nitrobenzene are analyzed by injection of a portion of the extract into an FIDGC. The dinitrotoluenes are analyzed by a separate injection into an ECDGC. Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and MDL that can be achieved under these conditions. Examples of the separations achieved by Column 1 are shown in Figures 1 and 2. Other packed or capillary (open-tubular) columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
12.2 Calibrate the system daily as described in Section 7.
12.3 If the internal standard calibration procedure is being used, the internal standard must be added to the same extract and mixed thoroughly immediately before injection into the gas chromatograph.
12.4 Inject 2 to 5 µL of the sample extract or standard into the gas chromatograph using the solvent-flush technique. 9 Smaller (1.0 µL) volumes may be injected if automatic devices are employed. Record the volume injected to the nearest 0.05 µL, the total extract volume, and the resulting peak size in area or peak height units.
12.5 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
12.6 If the response for a peak exceeds the working range of the system, dilute the extract and reanalyze.
12.7 If the measurement of the peak response is prevented by the presence of interferences, further cleanup is required.
13. Calculations13.1 Determine the concentration of individual compounds in the sample.
13.1.1 If the external standard calibration procedure is used, calculate the amount of material injected from the peak response using the calibration curve or calibration factor determined in Section 7.2.2. The concentration in the sample can be calculated from Equation 2.
Equation 2 where: A = Amount of material injected (ng). Vi = Volume of extract injected (µL). Vt = Volume of total extract (µL). Vs = Volume of water extracted (mL).13.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.3.2 and Equation 3.
Equation 3 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).13.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
14. Method Performance14.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Table 1 were obtained using reagent water. 10 Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
14.2 This method has been tested for linearity of spike recovery from reagent water and has been demonstrated to be applicable over the concentration range from 7 × MDL to 1000 × MDL. 10
14.3 This method was tested by 18 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 1.0 to 515 µg/L. 11 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. 40 CFR part 136, appendix B.
2. “Determination of Nitroaromatic Compounds and Isophorone in Industrial and Municipal Wastewaters,” EPA 600/ 4-82-024, National Technical Information Service, PB82-208398, Springfield, Virginia 22161, May 1982.
3. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia.
4. “Carcinogens - Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
5. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
7. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
8. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
9. Burke, J.A. “Gas Chromatography for Pesticide Residue Analysis; Some Practical Aspects,” Journal of the Association of Official Analytical Chemists, 48, 1037 (1965).
10. “Determination of Method Detection Limit and Analytical Curve for EPA Method 609 - Nitroaromatics and Isophorone,” Special letter report for EPA Contract 68-03-2624, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, June 1980.
11. “EPA Method Study 19, Method 609 (Nitroaromatics and Isophorone),” EPA 600/4-84-018, National Technical Information Service, PB84-176908, Springfield, Virginia 22161, March 1984.
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Method detection limit (µg/L) | ||
|---|---|---|---|---|
| Col. 1 | Col. 2 | ECDGC | FIDGC | |
| Nitrobenzene | 3.31 | 4.31 | 13.7 | 3.6 |
| 2,6-Dinitrotoluene | 3.52 | 4.75 | 0.01 | − |
| Isophorone | 4.49 | 5.72 | 15.7 | 5.7 |
| 2,4-Dinitrotoluene | 5.35 | 6.54 | 0.02 | − |
Column 1 conditions: Gas-Chrom Q (80/100 mesh) coated with 1.95% QF-1/1.5% OV-17 packed in a 1.2 m long × 2 mm or 4 mm ID glass column. A 2 mm ID column and nitrogen carrier gas at 44 mL/min flow rate were used when determining isophorone and nitrobenzene by FIDGC. The column temperature was held isothermal at 85 °C. A 4 mm ID column and 10% methane/90% argon carrier gas at 44 mL/min flow rate were used when determining the dinitrotoluenes by ECDGC. The column temperature was held isothermal at 145 °C.
Column 2 conditions: Gas-Chrom Q (80/100 mesh) coated with 3% OV-101 packed in a 3.0 m long × 2 mm or 4 mm ID glass column. A 2 mm ID column and nitrogen carrier gas at 44 mL/min flow rate were used when determining isophorone and nitrobenzene by FIDGC. The column temperature was held isothermal at 100 °C. A 4 mm ID column and 10% methane/90% argon carrier gas at 44 mL/min flow rate were used when determining the dinitrotoluenes by ECDGC. The column temperature was held isothermal at 150 °C.
Table 2 - QC Acceptance Criteria - Method 609
| Parameter | Test Conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps (%) |
|---|---|---|---|---|
| 2,4-Dinitrotoluene | 20 | 5.1 | 3.6-22.8 | 6-125 |
| 2,6-Dinitrotoluene | 20 | 4.8 | 3.8-23.0 | 8-126 |
| Isophorone | 100 | 32.3 | 8.0-100.0 | D-117 |
| Nitrobenzene | 100 | 33.3 | 25.7-100.0 | 6-118 |
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
D = Detected; result must be greater than zero.
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 609
| Parameter | Accuracy, as recovery, X′ (µg/L) | Single analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| 2,4-Dinitro- | |||
| toluene | 0.65C + 0.22 | 0.20X + 0.08 | 0.37X −0.07 |
| 2,6-Dinitro- | |||
| toluene | 0.66C + 0.20 | 0.19X + 0.06 | 0.36X −0.00 |
| Isophorone | 0.49C + 2.93 | 0.28X + 2.77 | 0.46X + 0.31 |
| Nitrobenzene | 0.60C + 2.00 | 0.25X + 2.53 | 0.37X −0.78 |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
 Method 610 -
Polynuclear Aromatic Hydrocarbons 1. Scope and Application
Method 610 -
Polynuclear Aromatic Hydrocarbons 1. Scope and Application
1.1 This method covers the determination of certain polynuclear aromatic hydrocarbons (PAH). The following parameters can be determined by this method:
| Parameter | STORET No. | CAS No. |
|---|---|---|
| Acenaphthene | 34205 | 83-32-9 |
| Acenaphthylene | 34200 | 208-96-8 |
| Anthracene | 34220 | 120-12-7 |
| Benzo(a)anthracene | 34526 | 56-55-3 |
| Benzo(a)pyrene | 34247 | 50-32-8 |
| Benzo(b)fluoranthene | 34230 | 205-99-2 |
| Benzo(ghi)perylene | 34521 | 191-24-2 |
| Benzo(k)fluoranthene | 34242 | 207-08-9 |
| Chrysene | 34320 | 218-01-9 |
| Dibenzo(a,h)anthracene | 34556 | 53-70-3 |
| Fluoranthene | 34376 | 206-44-0 |
| Fluorene | 34381 | 86-73-7 |
| Indeno(1,2,3-cd)pyrene | 34403 | 193-39-5 |
| Naphthalene | 34696 | 91-20-3 |
| Phenanthrene | 34461 | 85-01-8 |
| Pyrene | 34469 | 129-00-0 |
1.2 This is a chromatographic method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compounds above, compound identifications should be supported by at least one additional qualitative technique. Method 625 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for many of the parameters listed above, using the extract produced by this method.
1.3 This method provides for both high performance liquid chromatographic (HPLC) and gas chromatographic (GC) approaches for the determination of PAHs. The gas chromatographic procedure does not adequately resolve the following four pairs of compounds: Anthracene and phenanthrene; chrysene and benzo(a)anthracene; benzo(b)fluoranthene and benzo(k)fluoranthene; and dibenzo(a,h) anthracene and indeno (1,2,3-cd)pyrene. Unless the purpose for the analysis can be served by reporting the sum of an unresolved pair, the liquid chromatographic approach must be used for these compounds. The liquid chromatographic method does resolve all 16 of the PAHs listed.
1.4 The method detection limit (MDL, defined in Section 15.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.5 The sample extraction and concentration steps in this method are essentially the same as in Methods 606, 608, 609, 611, and 612. Thus, a single sample may be extracted to measure the parameters included in the scope of each of these methods. When cleanup is required, the concentration levels must be high enough to permit selecting aliquots, as necessary, to apply appropriate cleanup procedures. Selection of the aliquots must be made prior to the solvent exchange steps of this method. The analyst is allowed the latitude, under Sections 12 and 13, to select chromatographic conditions appropriate for the simultaneous measurement of combinations of these parameters.
1.6 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.7 This method is restricted to use by or under the supervision of analysts experienced in the use of HPLC and GC systems and in the interpretation of liquid and gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is extracted with methylene chloride using a separatory funnel. The methylene chloride extract is dried and concentrated to a volume of 10 mL or less. The extract is then separated by HPLC or GC. Ultraviolet (UV) and fluorescence detectors are used with HPLC to identify and measure the PAHs. A flame ionization detector is used with GC. 2
2.2 The method provides a silica gel column cleanup procedure to aid in the elimination of interferences that may be encountered.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardward that lead to discrete artifacts and/or elevated baselines in the chromatograms. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 3 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Some thermally stable materials, such as PCBs, may not be eliminated by this treatment. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Thorough rinsing with such solvents usually eliminates PCB interference. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to minimize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Matrix interferences may be caused by contaminants that are co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. The cleanup procedure in Section 11 can be used to overcome many of these interferences, but unique samples may require additional cleanup approaches to achieve the MDL listed in Table 1.
3.3 The extent of interferences that may be encountered using liquid chromatographic techniques has not been fully assessed. Although the HPLC conditions described allow for a unique resolution of the specific PAH compounds covered by this method, other PAH compounds may interfere.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method have not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 4-6 for the information of the analyst.
4.2 The following parameters covered by this method have been tentatively classified as known or suspected, human or mammalian carcinogens: benzo(a)anthracene, benzo(a)pyrene, and dibenzo(a,h)-anthracene. Primary standards of these toxic compounds should be prepared in a hood. A NIOSH/MESA approved toxic gas respirator should be worn when the analyst handles high concentrations of these toxic compounds.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1-L or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flow meter is required to collect flow proportional composites.
5.2 Glassware (All specifications are suggested. Catalog numbers are included for illustration only.):
5.2.1 Separatory funnel - 2-L, with Teflon stopcock.
5.2.2 Drying column - Chromatographic column, approximately 400 mm long × 19 mm ID, with coarse frit filter disc.
5.2.3 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes K-570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. Ground glass stopper is used to prevent evaporation of extracts.
5.2.4 Evaporative flask, Kuderna-Danish - 500-mL (Kontes K-570001-0500 or equivalent). Attach to concentrator tube with springs.
5.2.5 Snyder column, Kuderna-Danish - Three-ball macro (Kontes K-503000-0121 or equivalent).
5.2.6 Snyder column, Kuderna-Danish - Two-ball micro (Kontes K-569001-0219 or equivalent).
5.2.7 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.2.8 Chromatographic column - 250 mm long × 10 mm ID, with coarse frit filter disc at bottom and Teflon stopcock.
5.3 Boiling chips - Approximately 10/40 mesh. Heat to 400 °C for 30 min or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 Balance - Analytical, capable of accurately weighing 0.0001 g.
5.6 High performance liquid chromatograph (HPLC) - An analytical system complete with column supplies, high pressure syringes, detectors, and compatible strip-chart recorder. A data system is recommended for measuring peak areas and retention times.
5.6.1 Gradient pumping system - Constant flow.
5.6.2 Reverse phase column - HC-ODS Sil-X, 5 micron particle diameter, in a 25 cm × 2.6 mm ID stainless steel column (Perkin Elmer No. 089-0716 or equivalent). This column was used to develop the method performance statements in Section 15. Guidelines for the use of alternate column packings are provided in Section 12.2.
5.6.3 Detectors - Fluorescence and/or UV detectors. The fluorescence detector is used for excitation at 280 nm and emission greater than 389 nm cutoff (Corning 3-75 or equivalent). Fluorometers should have dispersive optics for excitation and can utilize either filter or dispersive optics at the emission detector. The UV detector is used at 254 nm and should be coupled to the fluorescence detector. These detectors were used to develop the method performance statements in Section 15. Guidelines for the use of alternate detectors are provided in Section 12.2.
5.7 Gas chromatograph - An analytical system complete with temperature programmable gas chromatograph suitable for on-column or splitless injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.7.1 Column - 1.8 m long × 2 mm ID glass, packed with 3% OV-17 on Chromosorb W-AW-DCMS (100/120 mesh) or equivalent. This column was used to develop the retention time data in Table 2. Guidelines for the use of alternate column packings are provided in Section 13.3.
5.7.2 Detector - Flame ionization detector. This detector has proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1), excluding the four pairs of unresolved compounds listed in Section 1.3. Guidelines for the use of alternate detectors are provided in Section 13.3.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.2 Sodium thiosulfate - (ACS) Granular.
6.3 Cyclohexane, methanol, acetone, methylene chloride, pentane - Pesticide quality or equivalent.
6.4 Acetonitrile - HPLC quality, distilled in glass.
6.5 Sodium sulfate - (ACS) Granular, anhydrous. Purify by heating at 400 °C for 4 h in a shallow tray.
6.6 Silica gel - 100/200 mesh, desiccant, Davison, grade-923 or equivalent. Before use, activate for at least 16 h at 130 °C in a shallow glass tray, loosely covered with foil.
6.7 Stock standard solutions (1.00 µg/µL) - Stock standard solutions can be prepared from pure standard materials or purchased as certified solutions.
6.7.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in acetonitrile and dilute to volume in a 10-mL volumetric flask. Larger volumes can be used at the convenience of the analyst. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.7.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store at 4 °C and protect from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.7.3 Stock standard solutions must be replaced after six months, or sooner if comparison with check standards indicates a problem.
6.8 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Establish liquid or gas chromatographic operating conditions equivalent to those given in Table 1 or 2. The chromatographic system can be calibrated using the external standard technique (Section 7.2) or the internal standard technique (Section 7.3).
7.2 External standard calibration procedure:
7.2.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with acetonitrile. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.2.2 Using injections of 5 to 25 µL for HPLC and 2 to 5 µL for GC, analyze each calibration standard according to Section 12 or 13, as appropriate. Tabulate peak height or area responses against the mass injected. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to amount injected (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.3 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask. To each calibration standard, add a known constant amount of one or more internal standards, and dilute to volume with acetonitrile. One of the standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.3.2 Using injections of 5 to 25 µL for HPLC and 2 to 5 µL for GC, analyze each calibration standard according to Section 12 or 13, as appropriate. Tabulate peak height or area responses against concentration for each compound and internal standard. Calculate response factors (RF) for each compound using Equation 1.
Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of the parameter to be measured (µg/L). If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.7.4 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of one or more calibration standards. If the response for any parameter varies from the predicted response by more than ±15%, the test must be repeated using a fresh calibration standard. Alternatively, a new calibration curve must be prepared for that compound.
7.5 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.4, 11.1, 12.2, and 13.3) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Before processing any samples the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest at the following concentrations in acetonitrile: 100 µg/mL of any of the six early-eluting PAHs (naphthalene, acenaphthylene, acenaphthene, fluorene, phenanthrene, and anthracene); 5 µg/mL of benzo(k)fluoranthene; and 10 µg/mL of any of the other PAHs. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at the test concentrations shown in Table 3 by adding 1.00 mL of QC check sample concentrate to each of four 1-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 3. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter.
Note:The large number of parameters in Table 3 present a substantial probability that one or more will fail at least one of the acceptance criteria when all parameters are analyzed.
8.2.6 When one or more of the parameters tested fail at least one of the acceptance criteria, the analyst must proceed according to Section 8.2.6.1 or 8.2.6.2.
8.2.6.1 Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.2.
8.2.6.2 Beginning with Section 8.2.2, repeat the test only for those parameters that failed to meet criteria. Repeated failure, however, will confirm a general problem with the measurement system. If this occurs, locate and correct the source of the problem and repeat the test for all compounds of interest beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at the test concentration in Section 8.2.2 or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determine background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any; or, if none, (2) the larger of either 5 times higher than the expected background concentration or the test concentration in Section 8.2.2.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100 (A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 3. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 7 If spiking was performed at a concentration lower than the test concentration in Section 8.2.2, the analyst must use either the QC acceptance criteria in Table 3, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 4, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 4, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T)±2.44(100 S′/T)%. 7
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the critiera must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory. If the entire list of parameters in Table 3 must be measured in the sample in Section 8.3, the probability that the analysis of a QC check standard will be required is high. In this case the QC check standard should be routinely analyzed with the spike sample.
8.4.1 Prepare the QC check standard by adding 1.0 mL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 1 L of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 3. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P -2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 8 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C from the time of collection until extraction. PAHs are known to be light sensitive; therefore, samples, extracts, and standards should be stored in amber or foil-wrapped bottles in order to minimize photolytic decomposition. Fill the sample bottles and, if residual chlorine is present, add 80 mg of sodium thiosulfate per liter of sample and mix well. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine. 9 Field test kits are available for this purpose.
9.3 All samples must be extracted within 7 days of collection and completely analyzed within 40 days of extraction. 2
10. Sample Extraction10.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel.
10.2 Add 60 mL of methylene chloride to the sample bottle, seal, and shake 30 s to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min. with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the methylene chloride extract in a 250-mL Erlenmeyer flask.
10.3 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.4 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator if the requirements of Section 8.2 are met.
10.5 Pour the combined extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the Erlenmeyer flask and column with 20 to 30 mL of methylene chloride to complete the quantitative transfer.
10.6 Add one or two clean boiling chips to the evaporative flask and attach a three-ball Snyder column. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
10.7 Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of methylene chloride. A 5-mL syringe is recommended for this operation. Stopper the concentrator tube and store refrigerated if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial and protected from light. If the sample extract requires no further cleanup, proceed with gas or liquid chromatographic analysis (Section 12 or 13). If the sample requires further cleanup, proceed to Section 11.
10.8 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1000-mL graduated cylinder. Record the sample volume to the nearest 5 mL.
11. Cleanup and Separation11.1 Cleanup procedures may not be necessary for a relatively clean sample matrix. If particular circumstances demand the use of a cleanup procedure, the analyst may use the procedure below or any other appropriate procedure. However, the analyst first must demonstrate that the requirements of Section 8.2 can be met using the methods as revised to incorporate the cleanup procedure.
11.2 Before the silica gel cleanup technique can be utilized, the extract solvent must be exchanged to cyclohexane. Add 1 to 10 mL of the sample extract (in methylene chloride) and a boiling chip to a clean K-D concentrator tube. Add 4 mL of cyclohexane and attach a two-ball micro-Snyder column. Prewet the column by adding 0.5 mL of methylene chloride to the top. Place the micro-K-D apparatus on a boiling (100 °C) water bath so that the concentrator tube is partially immersed in the hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete concentration in 5 to 10 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood. When the apparent volume of the liquid reaches 0.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min. Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with a minimum amount of cyclohexane. Adjust the extract volume to about 2 mL.
11.3 Silica gel column cleanup for PAHs:
11.3.1 Prepare a slurry of 10 g of activiated silica gel in methylene chloride and place this into a 10-mm ID chromatographic column. Tap the column to settle the silica gel and elute the methylene chloride. Add 1 to 2 cm of anhydrous sodium sulfate to the top of the silica gel.
11.3.2 Preelute the column with 40 mL of pentane. The rate for all elutions should be about 2 mL/min. Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, transfer the 2-mL cyclohexane sample extract onto the column using an additional 2 mL cyclohexane to complete the transfer. Just prior to exposure of the sodium sulfate layer to the air, add 25 mL of pentane and continue the elution of the column. Discard this pentane eluate.
11.3.3 Next, elute the column with 25 mL of methylene chloride/pentane (4 + 6)(V/V) into a 500-mL K-D flask equipped with a 10-mL concentrator tube. Concentrate the collected fraction to less than 10 mL as in Section 10.6. When the apparatus is cool, remove the Snyder column and rinse the flask and its lower joint with pentane. Proceed with HPLC or GC analysis.
12. High Performance Liquid Chromatography12.1 To the extract in the concentrator tube, add 4 mL of acetonitrile and a new boiling chip, then attach a two-ball micro-Snyder column. Concentrate the solvent as in Section 10.6, except set the water bath at 95 to 100 °C. When the apparatus is cool, remove the micro-Snyder column and rinse its lower joint into the concentrator tube with about 0.2 mL of acetonitrile. Adjust the extract volume to 1.0 mL.
12.2 Table 1 summarizes the recommended operating conditions for the HPLC. Included in this table are retention times, capacity factors, and MDL that can be achieved under these conditions. The UV detector is recommended for the determination of naphthalene, acenaphthylene, acenapthene, and fluorene and the fluorescence detector is recommended for the remaining PAHs. Examples of the separations achieved by this HPLC column are shown in Figures 1 and 2. Other HPLC columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
12.3 Calibrate the system daily as described in Section 7.
12.4 If the internal standard calibration procedure is being used, the internal standard must be added to the sample extract and mixed thoroughly immediately before injection into the instrument.
12.5 Inject 5 to 25 µL of the sample extract or standard into the HPLC using a high pressure syringe or a constant volume sample injection loop. Record the volume injected to the nearest 0.1 µL, and the resulting peak size in area or peak height units. Re-equilibrate the HPLC column at the initial gradient conditions for at least 10 min between injections.
12.6 Identify the parameters in the sample by comparing the retention time of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
12.7 If the response for a peak exceeds the working range of the system, dilute the extract with acetonitrile and reanalyze.
12.8 If the measurement of the peak response is prevented by the presence of interferences, further cleanup is required.
13. Gas Chromatography13.1 The packed column GC procedure will not resolve certain isomeric pairs as indicated in Section 1.3 and Table 2. The liquid chromatographic procedure (Section 12) must be used for these parameters.
13.2 To achieve maximum sensitivity with this method, the extract must be concentrated to 1.0 mL. Add a clean boiling chip to the methylene chloride extract in the concentrator tube. Attach a two-ball micro-Snyder column. Prewet the micro-Snyder column by adding about 0.5 mL of methylene chloride to the top. Place the micro-K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 5 to 10 min. At the proper rate of distillation the balls will actively chatter but the chambers will not flood. When the apparent volume of liquid reaches 0.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min. Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with a minimum amount of methylene chloride. Adjust the final volume to 1.0 mL and stopper the concentrator tube.
13.3 Table 2 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times that were obtained under these conditions. An example of the separations achieved by this column is shown in Figure 3. Other packed or capillary (open-tubular) columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
13.4 Calibrate the gas chromatographic system daily as described in Section 7.
13.5 If the internal standard calibration procedure is being used, the internal standard must be added to the sample extract and mixed thoroughly immediately before injection into the gas chromatograph.
13.6 Inject 2 to 5 µL of the sample extract or standard into the gas chromatograph using the solvent-flush technique. 10 Smaller (1.0 µL) volumes may be injected if automatic devices are employed. Record the volume injected to the nearest 0.05 µL, and the resulting peak size in area or peak height units.
13.7 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
13.8 If the response for a peak exceeds the working range of the system, dilute the extract and reanalyze.
13.9 If the measurement of the peak response is prevented by the presence of interferences, further cleanup is required.
14. Calculations14.1 Determine the concentration of individual compounds in the sample.
14.1.1 If the external standard calibration procedure is used, calculate the amount of material injected from the peak response using the calibration curve or calibration factor determined in Section 7.2.2. The concentration in the sample can be calculated from Equation 2.
Equation 2 where: A = Amount of material injected (ng). Vi = Volume of extract injected (µL). Vt = Volume of total extract (µL). Vs = Volume of water extracted (mL).13.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.3.2 and Equation 3.
Equation 3 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).14.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
15. Method Performance15.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Table 1 were obtained using reagent water. 11 Similar results were achieved using representative wastewaters. MDL for the GC approach were not determined. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
15.2 This method has been tested for linearity of spike recovery from reagent water and has been demonstrated to be applicable over the concentration range from 8 × MDL to 800 × MDL 11 with the following exception: benzo(ghi)perylene recovery at 80 × and 800 × MDL were low (35% and 45%, respectively).
15.3 This method was tested by 16 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 0.1 to 425 µg/L. 12 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 4.
References1. 40 CFR part 136, appendix B.
2. “Determination of Polynuclear Aromatic Hydrocarbons in Industrial and Municipal Wastewaters,” EPA 600/4-82-025, National Technical Information Service, PB82-258799, Springfield, Virginia 22161, June 1982.
3. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia.
4. “Carcinogens - Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
5. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
7. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
8. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
9. “Methods 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric, DPD) for Chlorine, Total Residual,” Methods for Chemical Analysis of Water and Wastes, EPA-600/4-79-020, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1979.
10. Burke, J.A. “Gas Chromatography for Pesticide Residue Analysis; Some Practical Aspects,” Journal of the Association of Official Analytical Chemists, 48, 1037 (1965).
11. Cole, T., Riggin, R., and Glaser, J. “Evaluation of Method Detection Limits and Analytical Curve for EPA Method 610 - PNAs,” International Symposium on Polynuclear Aromatic Hydrocarbons, 5th, Battelle's Columbus Laboratories, Columbus, Ohio (1980).
12. “EPA Method Study 20, Method 610 (PNA's),” EPA 600/4-84-063, National Technical Information Service, PB84-211614, Springfield, Virginia 22161, June 1984.
Table 1 - High Performance Liquid Chromatography Conditions and Method Detection Limits
| Parameter | Retention time (min) | Column capacity factor (k′) | Method detection limit (µg/L) a |
|---|---|---|---|
| Naphthalene | 16.6 | 12.2 | 1.8 |
| Acenaphthylene | 18.5 | 13.7 | 2.3 |
| Acenaphthene | 20.5 | 15.2 | 1.8 |
| Fluorene | 21.2 | 15.8 | 0.21 |
| Phenanthrene | 22.1 | 16.6 | 0.64 |
| Anthracene | 23.4 | 17.6 | 0.66 |
| Fluoranthene | 24.5 | 18.5 | 0.21 |
| Pyrene | 25.4 | 19.1 | 0.27 |
| Benzo(a)anthracene | 28.5 | 21.6 | 0.013 |
| Chrysene | 29.3 | 22.2 | 0.15 |
| Benzo(b)fluoranthene | 31.6 | 24.0 | 0.018 |
| Benzo(k)fluoranthene | 32.9 | 25.1 | 0.017 |
| Benzo(a)pyrene | 33.9 | 25.9 | 0.023 |
| Dibenzo(a,h)anthracene | 35.7 | 27.4 | 0.030 |
| Benzo(ghi)perylene | 36.3 | 27.8 | 0.076 |
| Indeno(1,2,3-cd)pyrene | 37.4 | 28.7 | 0.043 |
HPLC column conditions: Reverse phase HC-ODS Sil-X, 5 micron particle size, in a 25 cm × 2.6 mm ID stainless steel column. Isocratic elution for 5 min. using acetonitrile/water (4 + 6), then linear gradient elution to 100% acetonitrile over 25 min. at 0.5 mL/min flow rate. If columns having other internal diameters are used, the flow rate should be adjusted to maintain a linear velocity of 2 mm/sec.
a The MDL for naphthalene, acenaphthylene, acenaphthene, and fluorene were determined using a UV detector. All others were determined using a fluorescence detector.
Table 2 - Gas Chromatographic Conditions and Retention Times
| Parameter | Retention time (min) |
|---|---|
| Naphthalene | 4.5 |
| Acenaphthylene | 10.4 |
| Acenaphthene | 10.8 |
| Fluorene | 12.6 |
| Phenanthrene | 15.9 |
| Anthracene | 15.9 |
| Fluoranthene | 19.8 |
| Pyrene | 20.6 |
| Benzo(a)anthracene | 24.7 |
| Chrysene | 24.7 |
| Benzo(b)fluoranthene | 28.0 |
| Benzo(k)fluoranthene | 28.0 |
| Benzo(a)pyrene | 29.4 |
| Dibenzo(a,h)anthracene | 36.2 |
| Indeno(1,2,3-cd)pyrene | 36.2 |
| Benzo(ghi)perylene | 38.6 |
GC Column conditions: Chromosorb W-AW-DCMS (100/120 mesh) coated with 3% OV-17 packed in a 1.8 × 2 mm ID glass column with nitrogen carrier gas at 40 mL/min. flow rate. Column temperature was held at 100 °C for 4 min., then programmed at 8 °C/min. to a final hold at 280 °C.
Table 3 - QC Acceptance Criteria - Method 610
| Parameter | Test conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps (%) |
|---|---|---|---|---|
| Acenaphthene | 100 | 40.3 | D-105.7 | D-124 |
| Acenaphthylene | 100 | 45.1 | 22.1-112.1 | D-139 |
| Anthracene | 100 | 28.7 | 11.2-112.3 | D-126 |
| Benzo(a)anthracene | 10 | 4.0 | 3.1-11.6 | 12-135 |
| Benzo(a)pyrene | 10 | 4.0 | 0.2-11.0 | D-128 |
| Benzo(b)fluor-anthene | 10 | 3.1 | 1.8-13.8 | 6-150 |
| Benzo(ghi)perylene | 10 | 2.3 | D-10.7 | D-116 |
| Benzo(k)fluo-ranthene | 5 | 2.5 | D-7.0 | D-159 |
| Chrysene | 10 | 4.2 | D-17.5 | D-199 |
| Dibenzo(a,h)an-thracene | 10 | 2.0 | 0.3-10.0 | D-110 |
| Fluoranthene | 10 | 3.0 | 2.7-11.1 | 14-123 |
| Fluorene | 100 | 43.0 | D-119 | D-142 |
| Indeno(1,2,3-cd)pyrene | 10 | 3.0 | 1.2-10.0 | D-116 |
| Naphthalene | 100 | 40.7 | 21.5-100.0 | D-122 |
| Phenanthrene | 100 | 37.7 | 8.4-133.7 | D-155 |
| Pyrene | 10 | 3.4 | 1.4-12.1 | D-140 |
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
D = Detected; result must be greater than zero.
Note: These criteria are based directly upon the method performance data in Table 4. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 4.
Table 4 - Method Accuracy and Precision as Functions of Concentration - Method 610
| Parameter | Accuracy, as recovery, X′ (µg/L) | Single analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| Acenaphthene | 0.52C + 0.54 | 0.39X + 0.76 | 0.53X + 1.32 |
| Acenaphthylene | 0.69C − 1.89 | 0.36X + 0.29 | 0.42X + 0.52 |
| Anthracene | 0.63C − 1.26 | 0.23X + 1.16 | 0.41X + 0.45 |
| Benzo(a)anthracene | 0.73C + 0.05 | 0.28X + 0.04 | 0.34X + 0.02 |
| Benzo(a)pyrene | 0.56C + 0.01 | 0.38X − 0.01 | 0.53X − 0.01 |
| Benzo(b)fluoranthene | 0.78C + 0.01 | 0.21X + 0.01 | 0.38X − 0.00 |
| Benzo(ghi)perylene | 0.44C + 0.30 | 0.25X + 0.04 | 0.58X + 0.10 |
| Benzo(k)fluoranthene | 0.59C + 0.00 | 0.44X − 0.00 | 0.69X + 0.01 |
| Chrysene | 0.77C − 0.18 | 0.32X − 0.18 | 0.66X − 0.22 |
| Dibenzo(a,h)anthracene | 0.41C + 0.11 | 0.24X + 0.02 | 0.45X + 0.03 |
| Fluoranthene | 0.68C + 0.07 | 0.22X + 0.06 | 0.32X + 0.03 |
| Fluorene | 0.56C − 0.52 | 0.44X − 1.12 | 0.63X − 0.65 |
| Indeno(1,2,3-cd)pyrene | 0.54C + 0.06 | 0.29X + 0.02 | 0.42X + 0.01 |
| Naphthalene | 0.57C − 0.70 | 0.39X − 0.18 | 0.41X + 0.74 |
| Phenanthrene | 0.72C − 0.95 | 0.29X + 0.05 | 0.47X − 0.25 |
| Pyrene | 0.69C − 0.12 | 0.25X + 0.14 | 0.42X − 0.00 |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
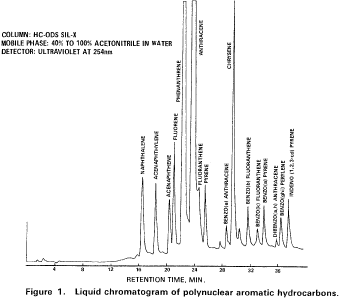
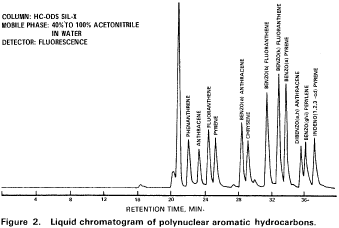
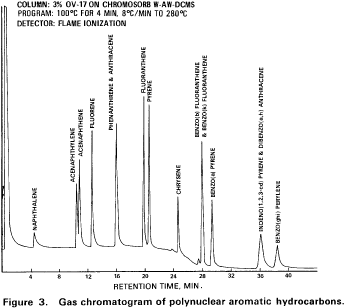 Method 611 -
Haloethers 1. Scope and Application
Method 611 -
Haloethers 1. Scope and Application
1.1 This method covers the determination of certain haloethers. The following parameters can be determined by this method:
| Parameter | STORET No. | CAS No. |
|---|---|---|
| Bis(2-chloroethyl) ether | 34273 | 111-44-4 |
| Bis(2-chloroethoxy) methane | 34278 | 111-91-1 |
| 2, 2′-oxybis (1-chloropropane) | 34283 | 108-60-1 |
| 4-Bromophenyl phenyl ether | 34636 | 101-55-3 |
| 4-Chlorophenyl phenyl ether | 34641 | 7005-72-3 |
1.2 This is a gas chromatographic (GC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compounds above, compound identifications should be supported by at least one additional qualitative technique. This method describes analytical conditions for a second gas chromatographic column that can be used to confirm measurements made with the primary column. Method 625 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for all of the parameters listed above, using the extract produced by this method.
1.3 The method detection limit (MDL, defined in Section 14.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.4 The sample extraction and concentration steps in this method are essentially the same as in Methods 606, 608, 609, and 612. Thus, a single sample may be extracted to measure the parameters included in the scope of each of these methods. When cleanup is required, the concentration levels must be high enough to permit selecting aliquots, as necessary, to apply appropriate cleanup procedures. The analyst is allowed the latitude, under Section 12, to select chromatographic conditions appropriate for the simultaneous measurement of combinations of these parameters.
1.5 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.6 This method is restricted to use by or under the supervision of analysts experienced in the use of a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is extracted with methylene chloride using a separatory funnel. The methylene chloride extract is dried and exchanged to hexane during concentration to a volume of 10 mL or less. The extract is separated by gas chromatography and the parameters are then measured with a halide specific detector. 2
2.2 The method provides a Florisil column cleanup procedure to aid in the elimination of interferences that may be encountered.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardware that lead to discrete artifacts and/or elevated baselines in gas chromatograms. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 3 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed be detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Some thermally stable materials, such a PCBs, may not be eliminated by this treatment. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Thorough rinsing with such solvents usually eliminates PCB interference. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to minimize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Matrix interferences may be caused by contaminants that are co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. The cleanup procedure in Section 11 can be used to overcome many of these interferences, but unique samples may require additional cleanup approaches to achieve the MDL listed in Table 1.
3.3 Dichlorobenzenes are known to coelute with haloethers under some gas chromatographic conditions. If these materials are present together in a sample, it may be necessary to analyze the extract with two different column packings to completely resolve all of the compounds.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 4-6 for the information of the analyst.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1-L or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flow meter is required to collect flow proportional composites.
5.2 Glassware (All specifications are suggested. Catalog numbers are included for illustration only.):
5.2.1 Separatory funnel - 2-L, with Teflon stopcock.
5.2.2 Drying column - Chromatographic column, approximately 400 mm long × 19 mm ID, with coarse frit filter disc.
5.2.3 Chromatographic column - 400 mm long × 19 mm ID, with Teflon stopcock and coarse frit filter disc at bottom (Kontes K-420540-0224 or equivalent).
5.2.4 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes K-570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. Ground glass stopper is used to prevent evaporation of extracts.
5.2.5 Evaporative flask, Kuderna-Danish - 500-mL (Kontes K-570001-0500 or equivalent). Attach to concentrator tube with springs.
5.2.6 Snyder column, Kuderna-Danish - Three-ball macro (Kontes K-503000-0121 or equivalent).
5.2.7 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.3 Boiling chips - Approximately 10/40 mesh. Heat to 400 °C for 30 min or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 Balance - Analytical, capable of accurately weighing 0.0001 g.
5.6 Gas chromatograph - An analytical system complete with temperature programmable gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.6.1 Column 1 - 1.8 m long × 2 mm ID glass, packed with 3% SP-1000 on Supelcoport (100/120 mesh) or equivalent. This column was used to develop the method performance statements in Section 14. Guidelines for the use of alternate column packings are provided in Section 12.1.
5.6.2 Column 2 - 1.8 m long × 2 mm ID glass, packed with 2,6-diphenylene oxide polymer (60/80 mesh), Tenax, or equivalent.
5.6.3 Detector - Halide specific detector: electrolytic conductivity or microcoulometric. These detectors have proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1). The Hall conductivity detector was used to develop the method performance statements in Section 14. Guidelines for the use of alternate detectors are provided in Section 12.1. Although less selective, an electron capture detector is an acceptable alternative.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.2 Sodium thiosulfate - (ACS) Granular.
6.3 Acetone, hexane, methanol, methylene chloride, petroleum ether (boiling range 30-60 °C) - Pesticide quality or equivalent.
6.4 Sodium sulfate - (ACS) Granular, anhydrous. Purify by heating at 400 °C for 4 h in a shallow tray.
6.5 Florisil - PR Grade (60/100 mesh). Purchase activated at 1250 °F and store in the dark in glass containers with ground glass stoppers or foil-lined screw caps. Before use, activate each batch at least 16 h at 130 °C in a foil-covered glass container and allow to cool.
6.6 Ethyl ether - Nanograde, redistilled in glass if necessary.
6.6.1 Ethyl ether must be shown to be free of peroxides before it is used as indicated by EM Laboratories Quant test strips. (Available from Scientific Products Co., Cat. No. P1126-8, and other suppliers.)
6.6.2 Procedures recommended for removal of peroxides are provided with the test strips. After cleanup, 20 mL of ethyl alcohol preservative must be added to each liter of ether.
6.7 Stock standard solutions (1.00 µg/µL) - Stock standard solutions can be prepared from pure standard materials or purchased as certified solutions.
6.7.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in acetone and dilute to volume in a 10-mL volumetric flask. Larger volumes can be used at the convenience of the analyst. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.7.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store at 4 °C and protect from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.7.3 Stock standard solutions must be replaced after six months, or sooner if comparison with check standards indicates a problem.
6.8 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Establish gas chromatographic operating conditions equivalent to those given in Table 1. The gas chromatographic system can be calibrated using the external standard technique (Section 7.2) or the internal standard technique (Section 7.3).
7.2 External standard calibration procedure:
7.2.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with hexane. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.2.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against the mass injected. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to amount injected (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.3 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask. To each calibration standard, add a known constant amount of one or more internal standards, and dilute to volume with hexane. One of the standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.3.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against concentration for each compound and internal standard. Calculate response factors (RF) for each compound using Equation 1.
Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of the parameter to be measured (µg/L). If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.7.4 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of one or more calibration standards. If the response for any parameter varies from the predicted response by more than ±15%, a new calibration curve must be prepared for that compound.
7.5 The cleanup procedure in Section 11 utilizes Florisil column chromatography. Florisil from different batches or sources may vary in adsorptive capacity. To standardize the amount of Florisil which is used, the use of lauric acid value 7 is suggested. The referenced procedure determines the adsorption from hexane solution of lauric acid (mg) per g of Florisil. The amount of Florisil to be used for each column is calculated by dividing 110 by this ratio and multiplying by 20 g.
7.6 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.4, 11.1, and 12.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Before processing any samples, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed, a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest at a concentration of 100 µg/mL in acetone. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at a concentration of 100 µg/L by adding 1.00 mL of QC check sample concentrate to each of four 1-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter. Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1. The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at 100 µg/L or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determine background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any; or, if none (2) the larger of either 5 times higher than the expected background concentration or 100 µg/L.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of each parameter. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100(A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 8 If spiking was performed at a concentration lower than 100 µg/L, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T)±2.44(100 S′/T)%. 8
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4 If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 1.0 m/L of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 1 L of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 2. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P -2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 9 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C from the time of collection until extraction. Fill the sample bottles and, if residual chlorine is present, add 80 mg of sodium thiosulfate per liter of sample and mix well. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine. 10 Field test kits are available for this purpose.
9.3 All samples must be extracted within 7 days of collection and completely analyzed within 40 days of extraction. 2
10. Sample Extraction10.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel.
10.2 Add 60 mL methylene chloride to the sample bottle, seal, and shake 30 s to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the methylene chloride extract in a 250-mL Erlenmeyer flask.
10.3 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.4 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator if the requirements of Section 8.2 are met.
10.5 Pour the combined extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the Erlenmeyer flask and column with 20 to 30 mL of methylene chloride to complete the quantitative transfer.
10.6 Add one or two clean boiling chips to the evaporative flask and attach a three-ball Snyder column. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
Note:Some of the haloethers are very volatile and significant losses will occur in concentration steps if care is not exercised. It is important to maintain a constant gentle evaporation rate and not to allow the liquid volume to fall below 1 to 2 mL before removing the K-D apparatus from the hot water bath.
10.7 Momentarily remove the Snyder column, add 50 mL of hexane and a new boiling chip, and reattach the Snyder column. Raise the temperature of the water bath to 85 to 90 °C. Concentrate the extract as in Section 10.6, except use hexane to prewet the column. The elapsed time of concentration should be 5 to 10 min.
10.8 Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of hexane. A 5-mL syringe is recommended for this operation. Stopper the concentrator tube and store refrigerated if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial. If the sample extract requires no further cleanup, proceed with gas chromatographic analysis (Section 12). If the sample requires further cleanup, proceed to Section 11.
10.9 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1000-mL graduated cylinder. Record the sample volume to the nearest 5 mL.
11. Cleanup and Separation11.1 Cleanup procedures may not be necessary for a relatively clean sample matrix. If particular circumstances demand the use of a cleanup procedure, the analyst may use the procedure below or any other appropriate procedure. However, the analyst first must demonstrate that the requirements of Section 8.2 can be met using the method as revised to incorporate the cleanup procedure.
11.2 Florisil column cleanup for haloethers:
11.2.1 Adjust the sample extract volume to 10 mL.
11.2.2 Place a weight of Florisil (nominally 20 g) predetermined by calibration (Section 7.5), into a chromatographic column. Tap the column to settle the Florisil and add 1 to 2 cm of anhydrous sodium sulfate to the top.
11.2.3 Preelute the column with 50 to 60 mL of petroleum ether. Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the sample extract onto the column by decantation and subsequent petroleum ether washings. Discard the eluate. Just prior to exposure of the sodium sulfate layer to the air, begin eluting the column with 300 mL of ethyl ether/petroleum ether (6 + 94) (V/V). Adjust the elution rate to approximately 5 mL/min and collect the eluate in a 500-mL K-D flask equipped with a 10-mL concentrator tube. This fraction should contain all of the haloethers.
11.2.4 Concentrate the fraction as in Section 10.6, except use hexane to prewet the column. When the apparatus is cool, remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with hexane. Adjust the volume of the cleaned up extract to 10 mL with hexane and analyze by gas chromatography (Section 12).
12. Gas Chromatography12.1 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and MDL that can be achieved under these conditions. Examples of the separations achieved by Columns 1 and 2 are shown in Figures 1 and 2, respectively. Other packed or capillary (open-tubular) columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
12.2 Calibrate the system daily as described in Section 7.
12.3 If the internal standard calibration procedure is being used, the internal standard must be added to the sample extract and mixed thoroughly immediately before injection into the gas chromatrograph.
12.4 Inject 2 to 5 µL of the sample extract or standard into the gas chromatograph using the solvent-flush technique. 11 Smaller (1.0 µL) volumes may be injected if automatic devices are employed. Record the volume injected to the nearest 0.05 µL, the total extract volume, and the resulting peak size in area or peak height units.
12.5 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weight heavily in the interpretation of chromatograms.
12.6 If the response for a peak exceeds the working range of the system, dilute the extract and reanalyze.
12.7 If the measurement of the peak response is prevented by the presence of interferences, further cleanup is required.
13. Calculations13.1 Determine the concentration of individual compounds in the sample.
13.1.1 If the external standard calibration procedure is used, calculate the amount of material injected from the peak response using the calibration curve or calibration factor determined in Section 7.2.2. The concentration in the sample can be calculated from Equation 2.
Equation 2 where: A = Amount of material injected (ng). Vi = Volume of extract injected (µL). Vt = Volume of total extract (µL). Vs = Volume of water extracted (mL).13.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.3.2 and Equation 3.
Equation 3 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).13.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
14. Method Performance14.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Table 1 were obtained using reagent water. 12 Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
14.2 This method has been tested for linearity of spike recovery from reagent water and has been demonstrated to be applicable over the concentration range from 4 × MDL to 1000 × MDL. 12
14.3 This method was tested by 20 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 1.0 to 626 µ/L. 12 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. 40 CFR part 136, appendix B.
2. “Determination of Haloethers in Industrial and Municipal Wastewaters,” EPA 600/4-81-062, National Technical Information Service, PB81-232290, Springfield, Virginia 22161, July 1981.
3. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constitutents,” American Society for Testing and Materials, Philadelphia.
4. “Carcinogens - Working Carcinogens, ” Department of Health, Education, and Welfare, Public Health Services, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
5. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
7. Mills., P.A. “Variation of Florisil Activity: Simple Method for Measuring Absorbent Capacity and Its Use in Standardizing Florisil Columns,” Journal of the Association of Official Analytical Chemists, 51, 29 (1968).
8. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
9. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
10. “Methods 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric, DPD) for Chlorine, Total Residual,” Methods for Chemical Analysis of Water and Wastes, EPA-600/4-79-020, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1979.
11. Burke, J.A. “Gas Chromatography for Pesticide Residue Analysis; Some Practical Aspects,” Journal of the Association of Official Analytical Chemists, 48, 1037 (1965).
12. “EPA Method Study 21, Method 611, Haloethers,” EPA 600/4-84-052, National Technical Information Service, PB84-205939, Springfield, Virginia 22161, June 1984.
Table 1 - Chromatographic Conditions and Methods Detection Limits
| Parameters | Retention time (min) | Method detection limit (µ/L) | |
|---|---|---|---|
| Column 1 | Column 2 | ||
| Bis(2-chloroisopropyl) ether | 8.4 | 9.7 | 0.8 |
| Bis(2-chloroethyl) ether | 9.3 | 9.1 | 0.3 |
| Bis(2-chloroethoxy) methane | 13.1 | 10.0 | 0.5 |
| 4-Chlorophenyl ether | 19.4 | 15.0 | 3.9 |
| 4-Bromophenyl phenyl ether | 21.2 | 16.2 | 2.3 |
Column 1 conditions: Supelcoport (100/120 mesh) coated with 3% SP-1000 packed in a 1.8 m long × 2 mm ID glass column with helium carrier gas at 40 mL/min. flow rate. Column temperature held at 60 °C for 2 min. after injection then programmed at 8 °C/min. to 230 °C and held for 4 min. Under these conditions the retention time for Aldrin is 22.6 min.
Column 2 conditions: Tenax-GC (60/80 mesh) packed in a 1.8 m long × 2mm ID glass column with helium carrier gas at 40 mL/min. flow rate. Column temperature held at 150 °C for 4 min. after injection then programmed at 16 °C/min. to 310 °C. Under these conditions the retention time for Aldrin is 18.4 min.
Table 2 - QC Acceptance Criteria - Method 611
| Parameter | Test conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps percent |
|---|---|---|---|---|
| Bis (2-chloroethyl)ether | 100 | 26.3 | 26.3-136.8 | 11-152 |
| Bis (2-chloroethoxy)methane | 100 | 25.7 | 27.3-115.0 | 12-128 |
| Bis (2-chloroisopropyl)ether | 100 | 32.7 | 26.4-147.0 | 9-165 |
| 4-Bromophenyl phenyl ether | 100 | 39.3 | 7.6-167.5 | D-189 |
| 4-Chlorophenyl phenyl ether | 100 | 30.7 | 15.4-152.5 | D-170 |
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
D = Detected; result must be greater than zero.
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 611
| Parameter | Accuracy, as recovery, X′ (µg/L) | Single analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| Bis(2-chloroethyl) ether | 0.81C + 0.54 | 0.19X + 0.28 | 0.35X + 0,36 |
| Bis(2-chloroethoxy) methane | 0.71C + 0.13 | 0.20X + 0.15 | 0.33X + 0.11 |
| Bis(2-chloroisopropyl) ether | 0.85C + 1.67 | 0.20X + 1.05 | 0.36X + 0.79 |
| 4-Bromophenyl phenyl ether | 0.85C + 2.55 | 0.25X + 0.21 | 0.47X + 0.37 |
| 4-Chlorophenyl phenyl ether | 0.82C + 1.97 | 0.18X + 2.13 | 0.41X + 0.55 |
X′ = Expected recovery for one or more measuremelts of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
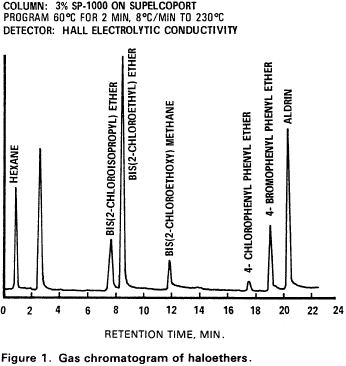
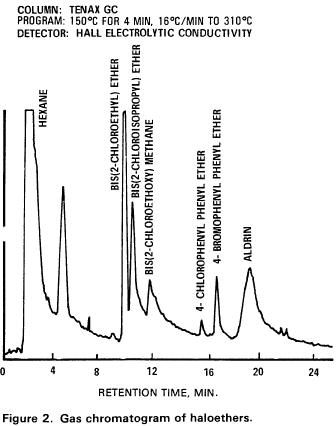 Method 612 -
Chlorinated Hydrocarbons 1. Scope and Application
Method 612 -
Chlorinated Hydrocarbons 1. Scope and Application
1.1 This method covers the determination of certain chlorinated hydrocarbons. The following parameters can be determined by this method:
| Parameter | STORET No. | CAS No. |
|---|---|---|
| 2-Chloronaphthalene | 34581 | 91-58-7 |
| 1,2-Dichlorobenzene | 34536 | 95-50-1 |
| 1,3-Dichlorobenzene | 34566 | 541-73-1 |
| 1,4-Dichlorobenzene | 34571 | 106-46-7 |
| Hexachlorobenzene | 39700 | 118-74-1 |
| Hexachlorobutadiene | 34391 | 87-68-3 |
| Hexachlorocyclopentadiene | 34386 | 77-47-4 |
| Hexachloroethane | 34396 | 67-72-1 |
| 1,2,4-Trichlorobenzene | 34551 | 120-82-1 |
1.2 This is a gas chromatographic (GC) method applicable to the determination of the compounds listed above in municipal and industrial discharges as provided under 40 CFR 136.1. When this method is used to analyze unfamiliar samples for any or all of the compounds above, compound identifications should be supported by at least one additional qualitative technique. This method describes a second gas chromatographic column that can be used to confirm measurements made with the primary column. Method 625 provides gas chromatograph/mass spectrometer (GC/MS) conditions appropriate for the qualitative and quantitative confirmation of results for all of the parameters listed above, using the extract produced by this method.
1.3 The method detection limit (MDL, defined in Section 14.1) 1 for each parameter is listed in Table 1. The MDL for a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.4 The sample extraction and concentration steps in this method are essentially the same as in Methods 606, 608, 609, and 611. Thus, a single sample may be extracted to measure the parameters included in the scope of each of these methods. When cleanup is required, the concentration levels must be high enough to permit selecting aliquots, as necessary, to apply appropriate cleanup procedures. The analyst is allowed the latitude, under Section 12, to select chromatographic conditions appropriate for the simultaneous measurement of combinations of these parameters.
1.5 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.6 This method is restricted to use by or under the supervision of analysts experienced in the use of a gas chromatograph and in the interpretation of gas chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is extracted with methylene chloride using a separatory funnel. The methylene chloride extract is dried and exchanged to hexane during concentration to a volume of 10 mL or less. The extract is separated by gas chromatography and the parameters are then measured with an electron capture detector. 2
2.2 The method provides a Florisil column cleanup procedure to aid in the elimination of interferences that may be encountered.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardware that lead to discrete artifacts and/or elevated baselines in gas chromatograms. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 3 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Some thermally stable materials, such as PCBs, may not be eliminated by this treatment. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Thorough rinsing with such solvents usually eliminates PCB interference. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to minimize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Matrix interferences may be caused by contaminants that are co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. The cleanup procedure in Section 11 can be used to overcome many of these interferences, but unique samples may require additional cleanup approaches to achieve the MDL listed in Table 1.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 4-6 for the information of the analyst.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1cL or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flow meter is required to collect flow proportional composites.
5.2 Glassware (All specifications are suggested. Catalog numbers are included for illustration only.):
5.2.1 Separatory funnel - 2-L, with Teflon stopcock.
5.2.2 Drying column - Chromatographic column, approximately 400 mm long × 19 mm ID, with coarse frit filter disc.
5.2.3 Chromatographic column - 300 long × 10 mm ID, with Teflon stopcock and coarse frit filter disc at bottom.
5.2.4 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes K-570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. Ground glass stopper is used to prevent evaporation of extracts.
5.2.5 Evaporative flask, Kuderna-Danish - 500-mL (Kontes K-570001-0500 or equivalent). Attach to concentrator tube with springs.
5.2.6 Snyder column, Kuderna-Danish - Three-ball macro (Kontes K-503000-0121 or equivalent).
5.2.7 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.3 Boiling chips - Approximately 10/40 mesh. Heat to 400 °C for 30 min or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 Balance - Analytical, capable of accurately weighing 0.0001 g.
5.6 Gas chromatograph - An analytical system complete with gas chromatograph suitable for on-column injection and all required accessories including syringes, analytical columns, gases, detector, and strip-chart recorder. A data system is recommended for measuring peak areas.
5.6.1 Column 1 - 1.8 m long × 2 mm ID glass, packed with 1% SP-1000 on Supelcoport (100/120 mesh) or equivalent. Guidelines for the use of alternate column packings are provide in Section 12.1.
5.6.2 Column 2 - 1.8 m long × 2 mm ID glass, packed with 1.5% OV-1/2.4% OV-225 on Supelcoport (80/100 mesh) or equivalent. This column was used to develop the method performance statements in Section 14.
5.6.3 Detector - Electron capture detector. This detector has proven effective in the analysis of wastewaters for the parameters listed in the scope (Section 1.1), and was used to develop the method performance statements in Section 14. Guidelines for the use of alternate detectors are provided in Section 12.1.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of the parameters of interest.
6.2 Acetone, hexane, isooctane, methanol, methylene chloride, petroleum ether (boiling range 30 to 60 °C) - Pesticide quality or equivalent.
6.3 Sodium sulfate - (ACS) Granular, anhydrous. Purify heating at 400 °C for 4 h in a shallow tray.
6.4 Florisil - PR grade (60/100 mesh). Purchase activated at 1250 °F and store in the dark in glass containers with ground glass stoppers or foil-lined screw caps. Before use, activate each batch at least 16 h at 130 °C in a foil-covered glass container and allow to cool.
6.5 Stock standard solution (1.00 µg/µL) - Stock standard solutions can be prepared from pure standard materials or purchased as certified solutions.
6.5.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in isooctane and dilute to volume in a 120-mL volumetric flask. Larger volumes can be used at the convenience of the analyst. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.5.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store at 4 °C and protect from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards from them.
6.5.3 Stock standard solutions must be replaced after six months, or sooner if comparision with check standards indicates a problem.
6.6 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Establish gas chromatographic operating conditions equivalent to those given in Table 1. The gas chromatographic system can be calibrated using the external standard technique (Section 7.2) or the internal standard technique (Section 7.3).
7.2 External standard calibration procedure:
7.2.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask and diluting to volume with isooctane. One of the external standards should be at a concentration near, but above, the MDL (Table 1) and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.2.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against the mass injected. The results can be used to prepare a calibration curve for each compound. Alternatively, if the ratio of response to amount injected (calibration factor) is a constant over the working range (<10% relative standard deviation, RSD), linearity through the origin can be assumed and the average ratio or calibration factor can be used in place of a calibration curve.
7.3 Internal standard calibration procedure - To use this approach, the analyst must select one or more internal standards that are similar in analytical behavior to the compounds of interest. The analyst must further demonstrate that the measurement of the internal standard is not affected by method or matrix interferences. Because of these limitations, no internal standard can be suggested that is applicable to all samples.
7.3.1 Prepare calibration standards at a minimum of three concentration levels for each parameter of interest by adding volumes of one or more stock standards to a volumetric flask. To each calibration standard, add a known constant amount of one or more internal standards, and dilute to volume with isooctane. One of the standards should be at a concentration near, but above, the MDL and the other concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the detector.
7.3.2 Using injections of 2 to 5 µL, analyze each calibration standard according to Section 12 and tabulate peak height or area responses against concentration for each compound and internal standard. Calculate response factors (RF) for each compound using Equation 1.
Equation 1 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of the parameter to be measured (µg/L). If the RF value over the working range is a constant (<10% RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.7.4 The working calibration curve, calibration factor, or RF must be verified on each working day by the measurement of one or more calibration standards. If the response for any parameter varies from the predicted response by more than ±15%, a new calibration curve must be prepared for that compound.
7.5 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When the results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.4, 11.1, and 12.1) to improve the separations or lower the cost of measurements. Each time such modification is made to the method, the analyst is required to repeat the procedure in Section 8.2.
8.1.3 Before processing any samples, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed, a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing each parameter of interest at the following concentrations in acetone: Hexachloro-substituted parameters, 10 µg/mL; any other chlorinated hydrocarbon, 100 µg/mL. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at the test concentrations shown in Table 2 by adding 1.00 mL of QC check sample concentrate to each of four 1-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for each parameter using the four results.
8.2.5 For each parameter compare s and X with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X for all parameters of interest meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, the system performance is unacceptable for that parameter.
Note:The large number of parameters in Table 2 presents a substantial probability that one or more will fail at least one of the acceptance criteria when all parameters are analyzed.
8.2.6 When one or more of the parameters tested fail at least one of the acceptance criteria, the analyst must proceed according to Section 8.2.6.1 or 8.2.6.2.
8.2.6.1 Locate and correct the source of the problem and repeat the test for all parameters of interest beginning with Section 8.2.2.
8.2.6.2 Beginning with Section 8.2.2, repeat the test only for those parameters that failed to meet criteria. Repeated failure, however, will confirm a general problem with the measurement system. If this occurs, locate and correct the source of the problem and repeat the test for all compounds of interest beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spike sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of a specific parameter in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of a specific parameter in the sample is not being checked against a limit specific to that parameter, the spike should be at the test concentration in Section 8.2.2 or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determine background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any; or, if none by (2) the larger of either 5 times higher than the expected background concentration or the test concentration in Section 8.2.2.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of each parameter. In necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentrations in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of each parameter. Calculate each percent recovery (P) as 100 (A−B)%/T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for each parameter with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 7 If spiking was performed at a concentration lower than the test concentration in Section 8.2.2, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of a parameter: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X ; (3) calculate the range for recovery at the spike concentration as (100 X′/T) ±2.44 (100 S′/T)%. 7
8.3.4 If any individual P falls outside the designated range for recovery, that parameter has failed the acceptance criteria. A check standard containing each parameter that failed the criteria must be analyzed as described in Section 8.4.
8.4. If any parameter fails the acceptance criteria for recovery in Section 8.3, a QC check standard containing each parameter that failed must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the number of parameters being simultaneously tested, the complexity of the sample matrix, and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 1.0 mL of QC check sample concentrate (Sections 8.2.1 or 8.3.2) to 1 L of reagent water. The QC check standard needs only to contain the parameters that failed criteria in the test in Section 8.3.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of each parameter. Calculate each percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) for each parameter with the corresponding QC acceptance criteria found in Table 2. Only parameters that failed the test in Section 8.3 need to be compared with these criteria. If the recovery of any such parameter falls outside the designated range, the laboratory performance for that parameter is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for that parameter in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P−2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each parameter on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. When doubt exists over the identification of a peak on the chromatogram, confirmatory techniques such as gas chromatography with a dissimilar column, specific element detector, or mass spectrometer must be used. Whenever possible, the laboratory should analyze standard reference materials and participate in relevent performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 8 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C from the time of collection until extraction.
9.3 All samples must be extracted within 7 days of collection and completely analyzed within 40 days of extraction. 2
10. Sample Extraction10.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel.
10.2 Add 60 mL of methylele chloride to the sample bottle, seal, and shake 30 s to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the methylene chloride extract in a 250-mL Erlenmeyer flask.
10.3 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.4 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator if the requirements of Section 8.2 are met.
10.5 Pour the combined extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the Erlenmeyer flask and column with 20 to 30 mL of methylene chloride to complete the quantitative transfer.
10.6 Add one or two clean boiling chips to the evaporative flask and attach a three-ball Snyder column. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 to 2 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
Note:The dichloribenzenes have a sufficiently high volatility that significant losses may occur in concentration steps if care is not exercised. It is important to maintain a constant gentle evaporation rate and not to allow the liquid volume to fall below 1 to 2 mL before removing the K-D apparatus from the hot water bath.
10.7 Momentarily remove the Snyder column, add 50 mL of hexane and a new boiling chip, and reattach the Snyder column. Raise the tempeature of the water bath to 85 to 90 °C. Concentrate the extract as in Section 10.6, except use hexane to prewet the column. The elapsed time of concentration should be 5 to 10 min.
10.8 Romove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of hexane. A 5-mL syringe is recommended for this operation. Stopper the concentrator tube and store refrigerated if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial. If the sample extract requires no further cleanup, proceed with gas chromatographic analysis (Section 12). If the sample requires further cleanup, proceed to Section 11.
10.9 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1000-mL graduated cylinder. Record the sample volume to the nearest 5 mL.
11. Cleanup and Separation11.1 Cleanup procedures may not be necessary for a relatively clean sample matrix. If particular circumstances demand the use of a cleanup procedure, the analyst may use the procedure below or any other appropriate procedure. However, the analyst first must demonstrate that the requirements of Section 8.2 can be met using the method as revised to incorporate the cleanup procedure.
11.2 Florisil column cleanup for chlorinated hydrocarbons:
11.2.1 Adjust the sample extract to 10 mL with hexane.
11.2.2 Place 12 g of Florisil into a chromatographic column. Tap the column to settle the Florisil and add 1 to 2 cm of anhydrous sodium sulfate to the top.
11.2.3 Preelute the column with 100 mL of petroleum ether. Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the sample extract onto the column by decantation and subsequent petroleum ether washings. Discard the eluate. Just prior to exposure of the sodium sulfate layer to the air, begin eluting the column with 200 mL of petroleum ether and collect the eluate in a 500-mL K-D flask equipped with a 10-mL concentrator tube. This fraction should contain all of the chlorinated hydrocarbons.
11.2.4 Concentrate the fraction as in Section 10.6, except use hexane to prewet the column. When the apparatus is cool, remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with hexane. Analyze by gas chromatography (Section 12).
12. Gas Chromatography12.1 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and MDL that can be achieved under these conditions. Examples of the separations achieved by Columl 2 are shown in Figures 1 and 2. Other packed or capillary (open-tubular) columns, chromatographic conditions, or detectors may be used if the requirements of Section 8.2 are met.
12.2 Calibrate the system daily as described in Section 7.
12.3 If the internal standard calibration procedure is being used, the internal standard must be added to the sample extract and mixed throughly immediately before injection into the gas chromatograph.
12.4 Inject 2 to 5 µL of the sample extract or standard into the gas chromatograph using the solvent-flush techlique. 9 Smaller (1.0 µL) volumes may be injected if automatic devices are employed. Record the volume injected to the nearest 0.05 µL, the total extract volume, and the resulting peak size in area or peak height units.
12.5 Identify the parameters in the sample by comparing the retention times of the peaks in the sample chromatogram with those of the peaks in standard chromatograms. The width of the retention time window used to make identifications should be based upon measurements of actual retention time variations of standards over the course of a day. Three times the standard deviation of a retention time for a compound can be used to calculate a suggested window size; however, the experience of the analyst should weigh heavily in the interpretation of chromatograms.
12.6 If the response for a peak exceeds the working range of the system, dilute the extract and reanalyze.
12.7 If the measurement of the peak response is prevented by the presence of interferences, further cleanup is required.
13. Calculations13.1 Determine the concentration of individual compounds in the sample.
13.1.1 If the external standard calibration procedure is used, calculate the amount of material injected from the peak response using the calibration curve or calibration factor determined in Section 7.2.2. The concentration in the sample can be calculated from Equation 2.
Equation 2 where: A = Amount of material injected (ng). Vi = Volume of extract injected (µL). Vt = Volume of total extract (µL). Vs = Volume of water extracted (mL).13.1.2 If the internal standard calibration procedure is used, calculate the concentration in the sample using the response factor (RF) determined in Section 7.3.2 and Equation 3.
Equation 3 where: As = Response for the parameter to be measured. Ais = Response for the internal standard. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).13.2 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
14. Method Performance14.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentrations listed in Table 1 were obtained using reagent water. 10 Similar results were achieved using representative wastewaters. The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
14.2 This method has been tested for linearity of spike recovery from reagent water and has been demonstrated to be applicable over the concentration range from 4 × MDL to 1000 × MDL. 10
14.3 This method was tested by 20 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 1.0 to 356 µg/L. 11 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. 40 CFR part 136, appendix B.
2. “Determination of Chlorinated Hydrocarbons In Industrial and Municipal Wastewaters, “EPA 6090/4-84-ABC, National Technical Information Service, PBXYZ, Springfield, Virginia, 22161 November 1984.
3. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia.
4. “Carcinogens - Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
5. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
7. Provost, L.P., and Elder, R.S. “Interpretation of Percent Recovery Data,”American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
8. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
9. Burke, J.A. “Gas Chromatography for Pesticide Residue Analysis; Some Practical Aspects,” Journal of the Association of Official Analytical Chemists, 48, 1037 (1965).
10. “Development of Detection Limits, EPA Method 612, Chlorinated Hydrocarbons,” Special letter report for EPA Contract 68-03-2625, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268.
11. “EPA Method Study Method 612 - Chlorinated Hydrocarbons,” EPA 600/4-84-039, National Technical Information Service, PB84-187772, Springfield, Virginia 22161, May 1984.
12. “Method Performance for Hexachlorocyclopentadiene by Method 612,” Memorandum from R. Slater, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, December 7, 1983.
Table 1 - Chromatographic Conditions and Method Detection Limits
| Parameter | Retention time (min) | Method detection limit (µg/L) | |
|---|---|---|---|
| Column 1 | Column 2 | ||
| 1,3-Dichlorobenzene | 4.5 | 6.8 | 1.19 |
| Hexachloroethane | 4.9 | 8.3 | 0.03 |
| 1,4-Dichlorobenzene | 5.2 | 7.6 | 1.34 |
| 1,2-Dichlorobenzene | 6.6 | 9.3 | 1.14 |
| Hexachlorobutadiene | 7.7 | 20.0 | 0.34 |
| 1,2,4-Trichlorobenzene | 15.5 | 22.3 | 0.05 |
| Hexachlorocyclopentadiene | nd | c 16.5 | 0.40 |
| 2-Chloronaphthalene | a 2.7 | b 3.6 | 0.94 |
| Hexachlorobenzene | a 5.6 | b 10.1 | 0.05 |
Column 1 conditions: Supelcoport (100/120 mesh) coated with 1% SP-1000 packed in a 1.8 m × 2 mm ID glass column with 5% methane/95% argon carrier gas at 25 mL/min. flow rate. Column temperature held isothermal at 65 °C, except where otherwise indicated.
Column 2 conditions: Supelcoport (80/100 mesh) coated with 1.5% OV-1/2.4% OV-225 packed in a 1.8 m × 2 mm ID glass column with 5% methane/95% argon carrier gas at 25 mL/min. flow rate. Column temperature held isothermal at 75 °C, except where otherwise indicated.
nd = Not determined.
a 150 °C column temperature.
b 165 °C column temperature.
c 100 °C column temperature.
Table 2 - QC Acceptance Criteria - Method 612
| Parameter | Test conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps (percent) |
|---|---|---|---|---|
| 2-Chloronaphthalene | 100 | 37.3 | 29.5-126.9 | 9-148 |
| 1,2-Dichlorobenzene | 100 | 28.3 | 23.5-145.1 | 9-160 |
| 1,3-Dichlorobenzene | 100 | 26.4 | 7.2-138.6 | D-150 |
| 1,4-Dichlorobenzene | 100 | 20.8 | 22.7-126.9 | 13-137 |
| Hexachlorobenzene | 10 | 2.4 | 2.6-14.8 | 15-159 |
| Hexachlorobutadiene | 10 | 2.2 | D-12.7 | D-139 |
| Hexachlorocyclopentadiene | 10 | 2.5 | D-10.4 | D-111 |
| Hexachloroethane | 10 | 3.3 | 2.4-12.3 | 8-139 |
| 1,2,4-Trichlorobenzene | 100 | 31.6 | 20.2-133.7 | 5-149 |
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
D = Detected; result must be greater than zero.
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 612
| Parameter | Acccuracy, as recovery, X′ (µg/L) | Single analyst precision, sr′ (µg/L) | Overall precision, S′ (µg/L) |
|---|---|---|---|
| 2-Chloronaphthalene | 0.75C + 3.21 | 0.28X −1.17 | 0.38X −1.39 |
| 1,2-Dichlorobenzene | 0.85C−0.70 | 0.22X −2.95 | 0.41X −3.92 |
| 1,3-Dichlorobenzene | 0.72C + 0.87 | 0.21X −1.03 | 0.49X −3.98 |
| 1,4-Dichlorobenzene | 0.72C + 2.80 | 0.16X −0.48 | 0.35X −0.57 |
| Hexachlorobenzene | 0.87C−0.02 | 0.14X + 0.07 | 0.36X −0.19 |
| Hexachlorobutadiene | 0.61C + 0.03 | 0.18X + 0.08 | 0.53X −0.12 |
| Hexachlorocyclopentadiene a | 0.47C | 0.24X | 0.50X |
| Hexachloroethane | 0.74C−0.02 | 0.23X + 0.07 | 0.36X −0.00 |
| 1,2,4-Trichlorobenzene | 0.76C + 0.98 | 0.23X −0.44 | 0.40X −1.37 |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
a Estimates based upon the performance in a single laboratory. 12
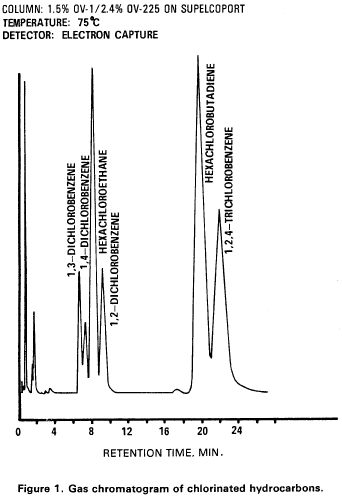
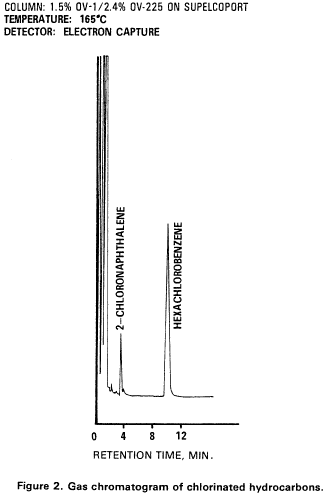 Method 613 -
2,3,7,8-Tetrachlorodibenzo-p-Dioxin 1. Scope and Application
Method 613 -
2,3,7,8-Tetrachlorodibenzo-p-Dioxin 1. Scope and Application
1.1 This method covers the determination of 2,3,7,8-tetrachlorodibenzo-p-dioxin (2,3,7,8-TCDD). The following parameter may be determined by this method:
| Parameter | STORET No. | GAS No. |
|---|---|---|
| 2,3,7,8-TCDD | 34675 | 1746-01-6 |
1.2 This is a gas chromatographic/mass spectrometer (GC/MS) method applicable to the determination of 2,3,7,8-TCDD in municipal and industrial discharges as provided under 40 CFR 136.1. Method 625 may be used to screen samples for 2,3,7,8-TCDD. When the screening test is positive, the final qualitative confirmation and quantification must be made using Method 613.
1.3 The method detection limit (MDL, defined in Section 14.1) 1 for 2,3,7,8-TCDD is listed in Table 1. The MDL for a specific wastewater may be different from that listed, depending upon the nature of interferences in the sample matrix.
1.4 Because of the extreme toxicity of this compound, the analyst must prevent exposure to himself, of to others, by materials knows or believed to contain 2,3,7,8-TCDD. Section 4 of this method contains guidelines and protocols that serve as minimum safe-handling standards in a limited-access laboratory.
1.5 Any modification of this method, beyond those expressly permitted, shall be considered as a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.6 This method is restricted to use by or under the supervision of analysts experienced in the use of a gas chromatograph/mass spectrometer and in the interpretation of mass spectra. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.
2. Summary of Method2.1 A measured volume of sample, approximately 1-L, is spiked with an internal standard of labeled 2,3,7,8-TCDD and extracted with methylene chloride using a separatory funnel. The methylene chloride extract is exchanged to hexane during concentration to a volume of 1.0 mL or less. The extract is then analyzed by capillary column GC/MS to separate and measure 2,3,7,8-TCDD. 2 3
2.2 The method provides selected column chromatographic cleanup proceudres to aid in the elimination of interferences that may be encountered.
3. Interferences3.1 Method interferences may be caused by contaminants in solvents, reagents, glassware, and other sample processing hardware that lead to discrete artifacts and/or elevated backgrounds at the masses (m/z) monitored. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis by running laboratory reagent blanks as described in Section 8.1.3.
3.1.1 Glassware must be scrupulously cleaned. 4 Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and distilled water. The glassware should then be drained dry, and heated in a muffle furnace at 400 °C for 15 to 30 min. Some thermally stable materials, such as PCBs, may not be eliminated by the treatment. Solvent rinses with acetone and pesticide quality hexane may be substituted for the muffle furnace heating. Thorough rinsing with such solvents usually eliminates PCB interference. Volumetric ware should not be heated in a muffle furnace. After drying and cooling, glassware should be sealed and stored in a clean environment to prevent any accumulation of dust or other contaminants. Store inverted or capped with aluminum foil.
3.1.2 The use of high purity reagents and solvents helps to mininmize interference problems. Purification of solvents by distillation in all-glass systems may be required.
3.2 Matrix interferences may be caused by contaminants that are coextracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. 2,3,7,8-TCDD is often associated with other interfering chlorinated compounds which are at concentrations several magnitudes higher than that of 2,3,7,8-TCDD. The cleanup producers in Section 11 can be used to overcome many of these interferences, but unique samples may require additional cleanup approaches 1 5-7 to eliminate false positives and achieve the MDL listed in Table 1.
3.3 The primary column, SP-2330 or equivalent, resolves 2,3,7,8-TCDD from the other 21 TCDD insomers. Positive results using any other gas chromatographic column must be confirmed using the primary column.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Additional references to laboratory safety are available and have been identified 8-10 for the information of the analyst. Benzene and 2,3,7,8-TCDD have been identified as suspected human or mammalian carcinogens.
4.2 Each laboratory must develop a strict safety program for handling 2,3,7,8-TCDD. The following laboratory practices are recommended:
4.2.1 Contamination of the laboratory will be minimized by conducting all manipulations in a hood.
4.2.2 The effluents of sample splitters for the gas chromatograph and roughing pumps on the GC/MS should pass through either a column of activated charcoal or be bubbled through a trap containing oil or high-boiling alcohols.
4.2.3 Liquid waste should be dissolved in methanol or ethanol and irradiated with ultraviolet light with a wavelength greater than 290 nm for several days. (Use F 40 BL lamps or equivalent). Analyze liquid wastes and dispose of the solutions when 2,3,7,8-TCDD can no longer be detected.
4.3 Dow Chemical U.S.A. has issued the following precautimns (revised November 1978) for safe handling of 2,3,7,8-TCDD in the laboratory:
4.3.1 The following statements on safe handling are as complete as possible on the basis of available toxicological information. The precautions for safe handling and use are necessarily general in nature since detailed, specific recommendations can be made only for the particular exposure and circumstances of each individual use. Inquiries about specific operations or uses may be addressed to the Dow Chemical Company. Assistance in evaluating the health hazards of particular plant conditions may be obtained from certain consulting laboratories and from State Departments of Health or of Labor, many of which have an industrial health service. 2,3,7,8-TCDD is extremely toxic to laboratory animals. However, it has been handled for years without injury in analytical and biological laboratories. Techniques used in handling radioactive and infectious materials are applicable to 2,3,7,8,-TCDD.
4.3.1.1 Protective equipment - Throw-away plastic gloves, apron or lab coat, safety glasses, and a lab hood adequate for radioactive work.
4.3.1.2 Training - Workers must be trained in the proper method of removing contaminated gloves and clothing without contacting the exterior surfaces.
4.3.1.3 Personal hygiene - Thorough washing of hands and forearms after each manipulation and before breaks (coffee, lunch, and shift).
4.3.1.4 Confinement - Isolated work area, posted with signs, segregated glassware and tools, plastic-backed absorbent paper on benchtops.
4.3.1.5 Waste - Good technique includes minimizing contaminated waste. Plastic bag liners should be used in waste cans. Janitors must be trained in the safe handling of waste.
4.3.1.6 Disposal of wastes - 2,3,7,8-TCDD decomposes above 800 °C. Low-level waste such as absorbent paper, tissues, animal remains, and plastic gloves may be burned in a good incinerator. Gross quantities (milligrams) should be packaged securely and disposed through commercial or governmental channels which are capable of handling high-level radioactive wastes or extremely toxic wastes. Liquids should be allowed to evaporate in a good hood and in a disposable container. Residues may then be handled as above.
4.3.1.7 Decontamination - For personal decontamination, use any mild soap with plenty of scrubbing action. For decontamination of glassware, tools, and surfaces, Chlorothene NU Solvent (Trademark of the Dow Chemical Company) is the least toxic solvent shown to be effective. Satisfactory cleaning may be accomplished by rinsing with Chlorothene, then washing with any detergent and water. Dishwater may be disposed to the sewer. It is prudent to minimize solvent wastes because they may require special disposal through commercial sources which are expensive.
4.3.1.8 Laundry - Clothing known to be contaminated should be disposed with the precautions described under Section 4.3.1.6. Lab coats or other clothing worn in 2,3,7,8-TCDD work areas may be laundered.
Clothing should be collected in plastic bags. Persons who convey the bags and launder the clothing should be advised of the hazard and trained in proper handling. The clothing may be put into a washer without contact if the launderer knows the problem. The washer should be run through a cycle before being used again for other clothing.
4.3.1.9 Wipe tests - A useful method of determining cleanliness of work surfaces and tools is to wipe the surface with a piece of filter paper. Extraction and analysis by gas chromatography can achieve a limit of sensitivity of 0.1 µg per wipe. Less than 1 µg of 2,3,7,8-TCDD per sample indicates acceptable cleanliness; anything higher warrants further cleaning. More than 10 µg on a wipe sample constitutes an acute hazard and requires prompt cleaning before further use of the equipment or work space. A high (>10 µg) 2,3,7,8-TCDD level indicates that unacceptable work practices have been employed in the past.
4.3.1.10 Inhalation - Any procedure that may produce airborne contamination must be done with good ventilation. Gross losses to a ventilation system must not be allowed. Handling of the dilute solutions normally used in analytical and animal work presents no inhalation hazards except in the case of an accident.
4.3.1.11 Accidents - Remove contaminated clothing immediately, taking precautions not to contaminate skin or other articles. Wash exposed skin vigorously and repeatedly until medical attention is obtained.
5. Apparatus and Materials5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - 1-L or 1-qt, amber glass, fitted with a screw cap lined with Teflon. Foil may be substituted for Teflon if the sample is not corrosive. If amber bottles are not available, protect samples from light. The bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - The sampler must incorporate glass sample containers for the collection of a minimum of 250 mL of sample. Sample containers must be kept refrigerated at 4 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, the compressible tubing should be thoroughly rinsed with methanol, followed by repeated rinsings with distilled water to minimize the potential for contamination of the sample. An integrating flow meter is required to collect flow proportional composites.
5.1.3 Clearly label all samples as “POISON” and ship according to U.S. Department of Transportation regulations.
5.2 Glassware (All specifications are suggested. Catalog numbers are included for illustration only.):
5.2.1 Separatory funnels - 2-L and 125-mL, with Teflon stopcock.
5.2.2 Concentrator tube, Kuderna-Danish - 10-mL, graduated (Kontes K-570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. Ground glass stopper is used to prevent evaporation of extracts.
5.2.3 Evaporative flask, Kuderna-Danish - 500-mL (Kontes K-570001-0500 or equivalent). Attach to concentrator tube with springs.
5.2.4 Snyder column, Kuderna-Danish - Three-ball macro (Kontes K-503000-0121 or equivalent).
5.2.5 Snyder column, Kuderna-Danish - Two-ball micro (Kontes K-569001-0219 or equivalent).
5.2.6 Vials - 10 to 15-mL, amber glass, with Teflon-lined screw cap.
5.2.7 Chromatographic column - 300 mm long × 10 mm ID, with Teflon stopcock and coarse frit filter disc at bottom.
5.2.8 Chromatographic column - 400 mm long × 11 mm ID, with Teflon stopcock and coarse frit filter disc at bottom.
5.3 Boiling chips - Approximately 10/40 mesh. Heat to 400 °C for 30 min or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 GC/MS system:
5.5.1 Gas chromatograph - An analytical system complete with a temperature programmable gas chromatograph and all required accessories including syringes, analytical columns, and gases. The injection port must be designed for capillary columns. Either split, splitless, or on-column injection techniques may be employed, as long as the requirements of Section 7.1.1 are achieved.
5.5.2 Column - 60 m long × 0.25 mm ID glass or fused silica, coated with SP-2330 (or equivalent) with a film thickness of 0.2 µm. Any equivalent column must resolve 2, 3, 7, 8-TCDD from the other 21 TCDD isomers. 16
5.5.3 Mass spectrometer - Either a low resolution mass spectrometer (LRMS) or a high resolution mass spectrometer (HRMS) may be used. The mass spectrometer must be equipped with a 70 V (nominal) ion source and be capable of aquiring m/z abundance data in real time selected ion monitoring (SIM) for groups of four or more masses.
5.5.4 GC/MS interface - Any GC to MS interface can be used that achieves the requirements of Section 7.1.1. GC to MS interfaces constructed of all glass or glass-lined materials are recommended. Glass surfaces can be deactivated by silanizing with dichlorodimethylsilane. To achieve maximum sensitivity, the exit end of the capillary column should be placed in the ion source. A short piece of fused silica capillary can be used as the interface to overcome problems associated with straightening the exit end of glass capillary columns.
5.5.5 The SIM data acquired during the chromatographic program is defined as the Selected Ion Current Profile (SICP). The SICP can be acquired under computer control or as a real time analog output. If computer control is used, there must be software available to plot the SICP and report peak height or area data for any m/z in the SICP between specified time or scan number limits.
5.6 Balance - Analytical, capable of accurately weighing 0.0001 g.
6. Reagents6.1 Reagent water - Reagent water is defined as a water in which an interferent is not observed at the MDL of 2, 3, 7, 8-TCDD.
6.2 Sodium hydroxide solution (10 N) - Dissolve 40 g of NaOH (ACS) in reagent water and dilute to 100 mL. Wash the solution with methylene chloride and hexane before use.
6.3 Sodium thiosulfate - (ACS) Granular.
6.4 Sulfuric acid - Concentrated (ACS, sp. gr. 1.84).
6.5 Acetone, methylene chloride, hexane, benzene, ortho-xylene, tetradecane - Pesticide quality or equivalent.
6.6 Sodium sulfate - (ACS) Granular, anhydrous. Purify by heating at 400 °C for 4 h in a shallow tray.
6.7 Alumina - Neutral, 80/200 mesh (Fisher Scientific Co., No. A-540 or equivalent). Before use, activate for 24 h at 130 °C in a foil-covered glass container.
6.8 Silica gel - High purity grade, 100/120 mesh (Fisher Scientific Co., No. S-679 or equivalent).
6.9 Stock standard solutions (1.00 µg/µL) - Stock standard solutimns can be prepared from pure standard materials or purchased as certified solutions. Acetone should be used as the solvent for spiking solutions; ortho-xylene is recommended for calibration standards for split injectors; and tetradecane is recommended for splitless or on-colum injectors. Analyze stock internal standards to verify the absence of native 2,3,7,8-TCDD.
6.9.1 Prepare stock standard solutions of 2,3,7,8-TCDD (mol wt 320) and either 37C14 2,3,7,8-TCDD (mol wt 328) or 13C112 2,3,7,8-TCDD (mol wt 332) in an isolated area by accurately weighing about 0.0100 g of pure material. Dissolve the material in pesticide quality solvent and dilute to volume in a 10-mL volumetric flask. When compound purity is assayed to be 96% or greater, the weight can be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards can be used at any concentration if they are certified by the manufacturer or by an independent source.
6.9.2 Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Store in an isolated refrigerator protected from light. Stock standard solutions should be checked frequently for signs of degradation or evaporation, especially just prior to preparing calibration standards or spiking solutions from them.
6.9.3 Stock standard solutions must be replaced after six months, or sooner if comparison with check standards indicates a problem.
6.10 Internal standard spiking solution (25 ng/mL) - Using stock standard solution, prepare a spiking solution in acetone of either 13 Cl12 or 37 Cl4 2,3,7,8-TCDD at a concentration of 25 ng/mL. (See Section 10.2)
6.11 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Establish gas chromatograhic operating conditions equivalent to those given in Table 1 and SIM conditions for the mass spectrometer as described in Section 12.2 The GC/MS system must be calibrated using the internal standard technique.
7.1.1 Using stock standards, prepare calibration standards that will allow measurement of relative response factors of at least three concentration ratios of 2,3,7,8-TCDD to internal standard. Each calibration standard must be prepared to contain the internal standard at a concentration of 25 ng/mL. If any interferences are contributed by the internal standard at m/z 320 and 322, its concentration may be reduced in the calibration standards and in the internal standard spiking solution (Section 6.10). One of the calibration standards should contain 2,3,7,8-TCDD at a concentration near, but above, the MDL and the other 2,3,7,8-TCDD concentrations should correspond to the expected range of concentrations found in real samples or should define the working range of the GC/MS system.
7.1.2 Using injections of 2 to 5 µL, analyze each calibration standardaccording to Section 12 and tabulate peak height or area response against the concentration of 2,3,7,8-TCDD and internal standard. Calculate response factors (RF) for 2,3,7,8-TCDD using Equation 1.
Equation 1 where: As = SIM response for 2,3,7,8-TCDD m/z 320. Ais = SIM response for the internal standard, m/z 332 for 13 C12 2,3,7,8-TCDD m/z 328 for 37 Cl4 2,3,7,8-TCDD. Cis = Concentration of the internal standard (µg/L). Cs = Concentration of 2,3,7,8-TCDD (µg/L). If the RF value over the working range is a constant (<10% relative standard deviation, RSD), the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to plot a calibration curve of response ratios, As/Ais, vs. RF.7.1.3 The working calibration curve or RF must be verified on each working day by the measurement of one or more 2,3,7,8-TCDD calibration standards. If the response for 2,3,7,8-TCDD varies from the predicted response by more than ±15%, the test must be repeated using a fresh calibration standard. Alternatively, a new calibration curve must be prepared.
7.2 Before using any cleanup procedure, the analyst must process a series of calibration standards through the procedure to validate elution patterns and the absence of interferences from the reagents.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records to document the quality of data that is generated. Ongoing data quality checks are compared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikes indicate atypical method performance, a quality control check standard must be analyzed to confirm that the measurements were performed in an in-control mode of operation.
8.1.1 The analyst must make an initial, one-time, demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 In recognition of advances that are occurring in chromatography, the analyst is permitted certain options (detailed in Sections 10.5, 11.1, and 12.1) to improve the separations or lower the cost of measurements. Each time such a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2
8.1.3 Before processing any samples, the analyst must analyze a reagent water blank to demonstrate that interferences from the analytical system and glassware are under control. Each time a set of samples is extracted or reagents are changed, a reagent water blank must be processed as a safeguard against laboratory contamination.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze a minimum of 10% of all samples with native 2,3,7,8-TCDD to monitor and evaluate laboratory data quality. This procedure is described in Section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through the analyses of quality control check standards that the operation of the measurement system is in control. This procedure is described in Section 8.4. The frequency of the check standard analyses is equivalent to 10% of all samples analyzed but may be reduced if spike recoveries from samples (Section 8.3) meet all specified quality control criteria.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is described in Section 8.5.
8.2 To establish the ability to generate acceptable accuracy and precision, the analyst must perform the following operations.
8.2.1 A quality control (QC) check sample concentrate is required containing 2,3,7,8-TCDD at a concentration of 0.100 µg/mL in acetone. The QC check sample concentrate must be obtained from the U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio, if available. If not available from that source, the QC check sample concentrate must be obtained from another external source. If not available from either source above, the QC check sample concentrate must be prepared by the laboratory using stock standards prepared independently from those used for calibration.
8.2.2 Using a pipet, prepare QC check samples at a concentration of 0.100 µg/L (100 ng/L) by adding 1.00 mL of QC check sample concentrate to each of four 1-L aliquots of reagent water.
8.2.3 Analyze the well-mixed QC check samples according to the method beginning in Section 10.
8.2.4 Calculate the average recovery (X ) in µg/L, and the standard deviation of the recovery (s) in µg/L, for 2,3,7,8-TCDD using the four results.
8.2.5 Compare s and (X ) with the corresponding acceptance criteria for precision and accuracy, respectively, found in Table 2. If s and X meet the acceptance criteria, the system performance is acceptable and analysis of actual samples can begin. If s exceeds the precision limit or X falls outside the range for accuracy, the system performance is unacceptable for 2,3,7,8-TCDD. Locate and correct the source of the problem and repeat the test beginning with Section 8.2.2.
8.3 The laboratory must, on an ongoing basis, spike at least 10% of the samples from each sample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.
8.3.1 The concentration of the spike in the sample should be determined as follows:
8.3.1.1 If, as in compliance monitoring, the concentration of 2,3,7,8-TCDD in the sample is being checked against a regulatory concentration limit, the spike should be at that limit or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.2 If the concentration of 2,3,7,8-TCDD in the sample is not being checked against a limit specific to that parameter, the spike should be at 0.100 µg/L or 1 to 5 times higher than the background concentration determined in Section 8.3.2, whichever concentration would be larger.
8.3.1.3 If it is impractical to determine background levels before spiking (e.g., maximum holding times will be exceeded), the spike concentration should be (1) the regulatory concentration limit, if any; or, if none (2) the larger of either 5 times higher than the expected background concentration or 0.100 µg/L.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of 2,3,7,8-TCDD. If necessary, prepare a new QC check sample concentrate (Section 8.2.1) appropriate for the background concentration in the sample. Spike a second sample aliquot with 1.0 mL of the QC check sample concentrate and analyze it to determine the concentration after spiking (A) of 2,3,7,8-TCDD. Calculate percent recovery (P) as 100(A−B)%T, where T is the known true value of the spike.
8.3.3 Compare the percent recovery (P) for 2,3,7,8-TCDD with the corresponding QC acceptance criteria found in Table 2. These acceptance criteria were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the analyst's spike to background ratio approaches 5:1. 11 If spiking was performed at a concentration lower than 0.100 µg/L, the analyst must use either the QC acceptance criteria in Table 2, or optional QC acceptance criteria calculated for the specific spike concentration. To calculate optional acceptance criteria for the recovery of 2,3,7,8-TCDD: (1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike concentration (T) for C; (2) calculate overall precision (S′) using the equation in Table 3, substituting X′ for X; (3) calculate the range for recovery at the spike concentration as (100 X′/T)±2.44(100 S′/T)%. 11
8.3.4 If the recovery of 2,3,7,8-TCDD falls outside the designated range for recovery, a check standard must be analyzed as described in Section 8.4.
8.4 If the recovery of 2,3,7,8-TCDD fails the acceptance criteria for recovery in Section 8.3, a QC check standard must be prepared and analyzed.
Note:The frequency for the required analysis of a QC check standard will depend upon the complexity of the sample matrix and the performance of the laboratory.
8.4.1 Prepare the QC check standard by adding 1.0 mL of QC check sample concentrate (Section 8.2.1 or 8.3.2) to 1 L of reagent water.
8.4.2 Analyze the QC check standard to determine the concentration measured (A) of 2,3,7,8-TCDD. Calculate the percent recovery (Ps) as 100 (A/T)%, where T is the true value of the standard concentration.
8.4.3 Compare the percent recovery (Ps) with the corresponding QC acceptance criteria found in Table 2. If the recovery of 2,3,7,8-TCDD falls outside the designated range, the laboratory performance is judged to be out of control, and the problem must be immediately identified and corrected. The analytical result for 2,3,7,8-TCDD in the unspiked sample is suspect and may not be reported for regulatory compliance purposes.
8.5 As part of the QC program for the laboratory, method accuracy for wastewater samples must be assessed and records must be maintained. After the analysis of five spiked wastewater samples as in Section 8.3, calculate the average percent recovery (P ) and the spandard deviation of the percent recovery (sp). Express the accuracy assessment as a percent recovery interval from P −2sp to P + 2sp. If P = 90% and sp = 10%, for example, the accuracy interval is expressed as 70-110%. Update the accuracy assessment on a regular basis (e.g. after each five to ten new accuracy measurements).
8.6 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of the environmental measurements. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Grab samples must be collected in glass containers. Conventional sampling practices 12 should be followed, except that the bottle must not be prerinsed with sample before collection. Composite samples should be collected in refrigerated glass containers in accordance with the requirements of the program. Automatic sampling equipment must be as free as possible of Tygon tubing and other potential sources of contamination.
9.2 All samples must be iced or refrigerated at 4 °C and protected from light from the time of collection until extraction. Fill the sample bottles and, if residual chlorine is present, add 80 mg of sodium thiosulfate per liter of sample and mix well. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine. 13 Field test kits are available for this purpose.
9.3 Label all samples and containers “POISON” and ship according to applicable U.S. Department of Transportation regulations.
9.4 All samples must be extracted within 7 days of collection and completely analyzed within 40 days of extraction. 2
10. Sample ExtractionCaution: When using this method to analyze for 2,3,7,8-TCDD, all of the following operations must be performed in a limited-access laboratory with the analyst wearing full protective covering for all exposed skin surfaces. See Section 4.2.
10.1 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into a 2-L separatory funnel.
10.2 Add 1.00 mL of internal standard spiking solution to the sample in the separatory funnel. If the final extract will be concentrated to a fixed volume below 1.00 mL (Section 12.3), only that volume of spiking solution should be added to the sample so that the final extract will contain 25 ng/mL of internal standard at the time of analysis.
10.3 Add 60 mL of methylene chloride to the sample bottle, seal, and shake 30 s to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for 2 min. with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 min. If the emulsion interface between layers is more than one-third the vmlume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool, centrifugation, or other physical methods. Collect the methylene chloride extract in a 250-mL Erlenmeyer flask.
10.4 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.5 Assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator if the requirements of Section 8.2 are met.
10.6 Pour the combined extract into the K-D concentrator. Rinse the Erlenmeyer flask with 20 to 30 mL of methylele chloride to complete the quantitative transfer.
10.7 Add one or two clean boiling chips to the evaporative flask and attach a three-ball Snyder column. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60 to 65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min.
10.8 Momentarily remove the Snyder column, add 50 mL of hexane and a new boiling chip, and reattach the Snyder column. Raise the temperature of the water bath to 85 to 90 °C. Concentrate the extract as in Section 10.7, except use hexane to prewet the column. Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of hexane. A 5-mL syringe is recommended for this operation. Set aside the K-D glassware for reuse in Section 10.14.
10.9 Pour the hexane extract from the concentrator tube into a 125-mL separatory funnel. Rinse the concentrator tube four times with 10-mL aliquots of hexane. Combine all rinses in the 125-mL separatory funnel.
10.10 Add 50 mL of sodium hydroxide solution to the funnel and shake for 30 to 60 s. Discard the aqueous phase.
10.11 Perform a second wash of the organic layer with 50 mL of reagent water. Discard the aqueous phase.
10.12 Wash the hexane layer with a least two 50-mL aliquots of concentrated sulfuric acid. Continue washing the hexane layer with 50-mL aliquots of concentrated sulfuric acid until the acid layer remains colorless. Discard all acid fractions.
10.13 Wash the hexane layer with two 50-mL aliquots of reagent water. Discard the aqueous phases.
10.14 Transfer the hexane extract into a 125-mL Erlenmeyer flask containing 1 to 2 g of anhydrous sodium sulfate. Swirl the flask for 30 s and decant the hexane extract into the reassembled K-D apparatus. Complete the quantitative transfer with two 10-mL hexane rinses of the Erlenmeyer flask.
10.15 Replace the one or two clean boiling chips and concentrate the extract to 6 to 10 mL as in Section 10.8.
10.16 Add a clean boiling chip to the concentrator tube and attach a two-ball micro-Snyder column. Prewet the column by adding about 1 mL of hexane to the top. Place the micro-K-D apparatus on the water bath so that the concentrator tube is partially immersed in the hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 5 to 10 min. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood. When the apparent volume of liquid reaches about 0.5 mL, remove the K-D apparatus and allow it to drain and cool for at least 10 min. Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with 0.2 mL of hexane.
Adjust the extract volume to 1.0 mL with hexane. Stopper the concentrator tube and store refrigerated and protected from light if further processing will not be performed immediately. If the extract will be stored longer than two days, it should be transferred to a Teflon-sealed screw-cap vial. If the sample extract requires no further cleanup, proceed with GC/MS analysis (Section 12). If the sample requires further cleanup, proceed to Section 11.
10.17 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to a 1000-mL graduated cylinder. Record the sample volume to the nearest 5 mL.
11. Cleanup and Separation11.1 Cleanup procedures may not be necessary for a relatively clean sample matrix. If particular circumstances demand the use of a cleanup procedure, the analyst may use either procedure below or any other appropriate procedure. 1 5-7 However, the analyst first must demonstrate that the requirements of Section 8.2 can be met using the method as revised to incorporate the cleanup procedure. Two cleanup column options are offered to the analyst in this section. The alumina column should be used first to overcome interferences. If background problems are still encountered, the silica gel column may be helpful.
11.2 Alumina column cleanup for 2,3,7,8-TCDD:
11.2.1 Fill a 300 mm long × 10 mm ID chromatographic column with activated alumina to the 150 mm level. Tap the column gently to settle the alumina and add 10 mm of anhydrous sodium sulfate to the top.
11.2.2 Preelute the column with 50 mL of hexane. Adjust the elution rate to 1 mL/min. Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the 1.0-mL sample extract onto the column using two 2-mL portions of hexane to complete the transfer.
11.2.3 Just prior to exposure of the sodium sulfate layer to the air, add 50 mL of 3% methylene chloride/95% hexane (V/V) and continue the elution of the column. Discard the eluate.
11.2.4 Next, elute the column with 50 mL of 20% methylene chloride/80% hexane (V/V) into a 500-mL K-D flask equipped with a 10-mL concentrator tube. Concentrate the collected fraction to 1.0 mL as in Section 10.16 and analyze by GC/MS (Section 12).
11.3 Silica gel column cleanup for 2,3,7,8-TCDD:
11.3.1 Fill a 400 mm long × 11 mm ID chromatmgraphic column with silica gel to the 300 mm level. Tap the column gently to settle the silica gel and add 10 mm of anhydrous sodium sulfate to the top.
11.3.2 Preelute the column with 50 mL of 20% benzene/80% hexane (V/V). Adjust the elution rate to 1 mL/min. Discard the eluate and just prior to exposure of the sodium sulfate layer to the air, quantitatively transfer the 1.0-mL sample extract onto the column using two 2-mL portions of 20% benzene/80% hexane to complete the transfer.
11.3.3 Just prior to exposure of the sodium sulfate layer to the air, add 40 mL of 20% benzene/80% hexane to the column. Collect the eluate in a clean 500-mL K-D flask equipped with a 10-mL concentrator tube. Concentrate the collected fraction to 1.0 mL as in Section 10.16 and analyze by GC/MS.
12. GC/MS Analysis12.1 Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and MDL that can be achieved under these conditions. Other capillary columns or chromatographic conditions may be used if the requirements of Sections 5.5.2 and 8.2 are met.
12.2 Analyze standards and samples with the mass spectrometer operating in the selected ion monitoring (SIM) mode using a dwell time to give at least seven points per peak. For LRMS, use masses at m/z 320, 322, and 257 for 2,3,7,8-TCDD and either m/z 328 for 37Cl4 2,3,7,8-TCDD or m/z 332 for 13C12 2,3,7,8-TCDD. For HRMS, use masses at m/z 319.8965 and 321.8936 for 2,3,7,8-TCDD and either m/z 327.8847 for 37Cl4 2,3,7,8-TCDD or m/z 331.9367 for 13C12 2,3,7,8-TCDD.
12.3 If lower detection limits are required, the extract may be carefully evaporated to dryness under a gentle stream of nitrogen with the concentrator tube in a water bath at about 40 °C. Conduct this operation immediately before GC/MS analysis. Redissolve the extract in the desired final volume of ortho-xylene or tetradecane.
12.4 Calibrate the system daily as described in Section 7.
12.5 Inject 2 to 5 µL of the sample extract into the gas chromatograph. The volume of calibration standard injected must be measured, or be the same as all sample injection volumes.
12.6 The presence of 2,3,7,8-TCDD is qualitatively confirmed if all of the following criteria are achieved:
12.6.1 The gas chromatographic column must resolve 2,3,7,8-TCDD from the other 21 TCDD isomers.
12.6.2 The masses for native 2,3,7,8-TCDD (LRMS-m/z 320, 322, and 257 and HRMS-m/z 320 and 322) and labeled 2,3,7,8-TCDD (m/z 328 or 332) must exhibit a simultaneous maximum at a retention time that matches that of native 2,3,7,8-TCDD in the calibration standard, with the performance specifications of the analytical system.
12.6.3 The chlorine isotope ratio at m/z 320 and m/z 322 must agree to within±10% of that in the calibration standard.
12.6.4 The signal of all peaks must be greater than 2.5 times the noise level.
12.7 For quantitation, measure the response of the m/z 320 peak for 2,3,7,8-TCDD and the m/z 332 peak for 13C12 2,3,7,8-TCDD or the m/z 328 peak for 37Cl4 2,3,7,8-TCDD.
12.8 Co-eluting impurities are suspected if all criteria are achieved except those in Section 12.6.3. In this case, another SIM analysis using masses at m/z 257, 259, 320 and either m/a 328 or m/z 322 can be performed. The masses at m/z 257 and m/z 259 are indicative of the loss of one chlorine and one carbonyl group from 2,3,7,8-TCDD. If masses m/z 257 and m/z 259 give a chlorine isotope ratio that agrees to within ±10% of the same cluster in the calibration standards, then the presence of TCDD can be confirmed. Co-eluting DDD, DDE, and PCB residues can be confirmed, but will require another injection using the appropriate SIM masses or full repetitive mass scans. If the response for 37Cl4 2,3,7,8-TCDD at m/z 328 is too large, PCB contamination is suspected and can be confirmed by examining the response at both m/z 326 and m/z 328. The 37Cl4 2,3,7,8-TCDD internal standard gives negligible response at m/z 326. These pesticide residues can be removed using the alumina column cleanup procedure.
12.9 If broad background interference restricts the sensitivity of the GC/MS analysis, the analyst should employ additional cleanup procedures and reanalyze by GC/MS.
12.10 In those circumstances where these procedures do not yield a definitive conclusion, the use of high resolution mass spectrometry is suggested. 5
13. Calculations13.1 Calculate the concentration of 2,3,7,8-TCDD in the sample using the response factor (RF) determined in Section 7.1.2 and Equation 2.
Equation 2 where: As = SIM response for 2,3,7,8-TCDD at m/z 320. Ais = SIM response for the internal standard at m/z 328 or 332. Is = Amount of internal standard added to each extract (µg). Vo = Volume of water extracted (L).13.2 For each sample, calculate the percent recovery of the internal standard by comparing the area of the m/z peak measured in the sample to the area of the same peak in the calibration standard. If the recovery is below 50%, the analyst should review all aspects of his analytical technique.
13.3 Report results in µg/L without correction for recovery data. All QC data obtained should be reported with the sample results.
14. Method Performance14.1 The method detection limit (MDL) is defined as the minimum concentration of a substance that can be measured and reported with 99% confidence that the value is above zero. 1 The MDL concentration listed in Table 1 was obtained using reagent water. 14 The MDL actually achieved in a given analysis will vary depending on instrument sensitivity and matrix effects.
14.2 This method was tested by 11 laboratories using reagent water, drinking water, surface water, and three industrial wastewaters spiked at six concentrations over the range 0.02 to 0.20 µg/L. 15 Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the parameter and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 3.
References1. 40 CFR part 136, appendix B.
2. “Determination of TCDD in Industrial and Municipal Wastewaters,” EPA 600/4-82-028, National Technical Information Service, PB82-196882, Springfield, Virginia 22161, April 1982.
3. Buser, H.R., and Rappe, C. “High Resolution Gas Chromatography of the 22 Tetrachlorodibenzo-p-dioxin Isomers,” Analytical Chemistry, 52, 2257 (1980).
4. ASTM Annual Book of Standards, Part 31, D3694-78. “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia.
5. Harless, R. L., Oswald, E. O., and Wilkinson, M. K. “Sample Preparation and Gas Chromatography/Mass Spectrometry Determination of 2,3,7,8-Tetrachlorodibenzo-p-dioxin,” Analytical Chemistry, 52, 1239 (1980).
6. Lamparski, L. L., and Nestrick, T. J. “Determination of Tetra-, Hepta-, and Octachlorodibenzo-p-dioxin Isomers in Particulate Samples at Parts per Trillion Levels,” Analytical Chemistry, 52, 2045 (1980).
7. Longhorst, M. L., and Shadoff, L. A. “Determination of Parts-per-Trillion Concentrations of Tetra-, Hexa-, and Octachlorodibenzo-p-dioxins in Human Milk,” Analytical Chemistry, 52, 2037 (1980).
8. “Carcinogens - Working with Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
9. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occuptional Safety and Health Administration, OSHA 2206 (Revised, January 1976).
10. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
11. Provost, L. P., and Elder, R. S., “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in Section 8.3.3 is two times the value 1.22 derived in this report.)
12. ASTM Annual Book of Standards, Part 31, D3370-76, “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia.
13. “Methods, 330.4 (Titrimetric, DPD-FAS) and 330.5 (Spectrophotometric DPD) for Chlorine, Total Residual,” Methods for Chemical Analysis of Water and Wastes, EPA-600/4-79-020, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1979.
14. Wong, A.S. et al. “The Determination of 2,3,7,8-TCDD in Industrial and Municipal Wastewaters, Method 613, Part 1 - Development and Detection Limits,” G. Choudhay, L. Keith, and C. Ruppe, ed., Butterworth Inc., (1983).
15. “EPA Method Study 26, Method 613: 2,3,7,8-Tetrachlorodibenzo-p-dioxin,” EPA 600/4-84-037, National Technical Information Service, PB84-188879, Springfield, Virginia 22161, May 1984.
Table 1 - Chromatographic Conditions and Method Detection Limit
| Parameter | Retention time (min) | Method detection limit (µg/L) |
|---|---|---|
| 2,3,7,8-TCDD | 13.1 | 0.002 |
Column conditions: SP-2330 coated on a 60 m long × 0.25 mm ID glass column with hydrogen carrier gas at 40 cm/sec linear velocity, splitless injection using tetradecane. Column temperature held isothermal at 200 °C for 1 min, then programmed at 8 °C/min to 250 °C and held. Use of helium carrier gas will approximately double the retention time.
Table 2 - QC Acceptance Criteria - Method 613
| Parameter | Test conc. (µg/L) | Limit for s (µg/L) | Range for X (µg/L) | Range for P, Ps (%) |
|---|---|---|---|---|
| 2,3,7,8-TCDD | 0.100 | 0.0276 | 0.0523-0.1226 | 45-129 |
s = Standard deviation of four recovery measurements, in µg/L (Section 8.2.4).
X = Average recovery for four recovery measurements, in µg/L (Section 8.2.4).
P, Ps = Percent recovery measured (Section 8.3.2, Section 8.4.2).
Note: These criteria are based directly upon the method performance data in Table 3. Where necessary, the limits for recovery have been broadened to assure applicability of the limits to concentrations below those used to develop Table 3.
Table 3 - Method Accuracy and Precision as Functions of Concentration - Method 613
| Parameter | Accuracy, as recovery, X″ (µg/L) | Single analyst, precision, sr″ (µ/L) | Overall precision, S″ (µ/g/L) |
|---|---|---|---|
| 2,3,7,8-TCDD | 0.86C + 0.00145 | 0.13X + 0.00129 | 0.19X + 0.00028 |
X′ = Expected recovery for one or more measurements. of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X , in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X , in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
1.1 This method is for determination of purgeable organic pollutants in industrial discharges and other environmental samples by gas chromatography combined with mass spectrometry (GC/MS), as provided under 40 CFR 136.1. This revision is based on previous protocols (References 1 - 3), on the revision promulgated October 26, 1984, and on an interlaboratory method validation study (Reference 4). Although this method was validated through an interlaboratory study conducted in the early 1980s, the fundamental chemistry principles used in this method remain sound and continue to apply.
1.2 The analytes that may be qualitatively and quantitatively determined using this method and their CAS Registry numbers are listed in Table 1. The method may be extended to determine the analytes listed in Table 2; however, poor purging efficiency or gas chromatography of some of these analytes may make quantitative determination difficult. For example, an elevated temperature may be required to purge some analytes from water. If an elevated temperature is used, calibration and all quality control (QC) tests must be performed at the elevated temperature. EPA encourages the use of this method to determine additional compounds amenable to purge-and-trap GC/MS.
1.3 The large number of analytes in Tables 1 and 2 of this method makes testing difficult if all analytes are determined simultaneously. Therefore, it is necessary to determine and perform QC tests for “analytes of interest” only. Analytes of interest are those required to be determined by a regulatory/control authority or in a permit, or by a client. If a list of analytes is not specified, the analytes in Table 1 must be determined, at a minimum, and QC testing must be performed for these analytes. The analytes in Table 1 and some of the analytes in Table 2 have been identified as Toxic Pollutants (40 CFR 401.15), expanded to a list of Priority Pollutants (40 CFR part 423, appendix A).
1.4 Method detection limits (MDLs; Reference 5) for the analytes in Table 1 are listed in that table. These MDLs were determined in reagent water (Reference 6). Advances in analytical technology, particularly the use of capillary (open-tubular) columns, allowed laboratories to routinely achieve MDLs for the analytes in this method that are 2-10 times lower than those in the version promulgated in 1984. The MDL for a specific wastewater may differ from those listed, depending on the nature of interferences in the sample matrix.
1.4.1 EPA has promulgated this method at 40 CFR part 136 for use in wastewater compliance monitoring under the National Pollutant Discharge Elimination System (NPDES). The data reporting practices described in section 13.2 are focused on such monitoring needs and may not be relevant to other uses of the method.
1.4.2 This method includes “reporting limits” based on EPA's “minimum level” (ML) concept (see the glossary in section 20). Table 1 contains MDL values and ML values for many of the analytes. The MDL for an analyte in a specific wastewater may differ from that listed in Table 1, depending upon the nature of interferences in the sample matrix.
1.5 This method is performance-based. It may be modified to improve performance (e.g., to overcome interferences or improve the accuracy of results) provided all performance requirements are met.
1.5.1 Examples of allowed method modifications are described at 40 CFR 136.6. Other examples of allowed modifications specific to this method are described in section 8.1.2.
1.5.2 Any modification beyond those expressly allowed at 40 CFR 136.6 or in section 8.1.2 of this method shall be considered a major modification that is subject to application and approval of an alternate test procedure under 40 CFR 136.4 and 136.5.
1.5.3 For regulatory compliance, any modification must be demonstrated to produce results equivalent or superior to results produced by this method when applied to relevant wastewaters (section 8.3).
1.6 This method is restricted to use by or under the supervision of analysts experienced in the operation of a purge-and-trap system and a gas chromatograph/mass spectrometer and in the interpretation of mass spectra. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure in section 8.2.
1.7 Terms and units of measure used in this method are given in the glossary at the end of the method.
2. Summary of Method2.1 A gas is bubbled through a measured volume of water in a specially-designed purging chamber. The purgeables are efficiently transferred from the aqueous phase to the vapor phase. The vapor is swept through a sorbent trap where the purgeables are trapped. After purging is completed, the trap is heated and backflushed with the gas to desorb the purgeables onto a gas chromatographic column. The column is temperature programmed to separate the purgeables which are then detected with a mass spectrometer.
2.2 Different sample sizes in the range of 5-25 mL are allowed in order to meet differing sensitivity requirements. Calibration and QC samples must have the same volume as field samples.
3. Interferences3.1 Impurities in the purge gas, organic compounds outgassing from the plumbing ahead of the trap, and solvent vapors in the laboratory account for the majority of contamination problems. The analytical system must be demonstrated to be free from contamination under the conditions of the analysis by analyzing blanks initially and with each analytical batch (samples analyzed on a given 12-hour shift, to a maximum of 20 samples), as described in Section 8.5. Fluoropolymer tubing, fittings, and thread sealant should be used to avoid contamination.
3.2 Samples can be contaminated by diffusion of volatile organics (particularly fluorocarbons and methylene chloride) through the septum seal into the sample during shipment and storage. Protect samples from sources of volatiles during collection, shipment, and storage. A reagent water field blank carried through sampling and analysis can serve as a check on such contamination.
3.3 Contamination by carry-over can occur whenever high level and low level samples are analyzed sequentially. To reduce the potential for carry-over, the purging device and sample syringe must be rinsed with reagent water between sample analyses. Whenever an unusually concentrated sample is encountered, it should be followed by an analysis of a blank to check for cross contamination. For samples containing large amounts of water-soluble materials, suspended solids, high boiling compounds or high purgeable levels, it may be necessary to wash the purging device with a detergent solution, rinse it with distilled water, and then dry it in a 105 °C oven between analyses. The trap and other parts of the system are also subject to contamination; therefore, frequent bakeout and purging of the entire system may be required. Screening samples at high dilution may prevent introduction of contaminants into the system.
4. Safety4.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of safety data sheets (SDSs, OSHA, 29 CFR 1910.1200(g)) should also be made available to all personnel involved in sample handling and chemical analysis. Additional references to laboratory safety are available and have been identified (References 7-9) for the information of the analyst.
4.2. The following analytes covered by this method have been tentatively classified as known or suspected human or mammalian carcinogens: Benzene; carbon tetrachloride; chloroform; 1,4-dichlorobenzene; 1,2-dichloroethane; 1,2-dichloropropane; methylene chloride; tetrachloroethylene; trichloroethylene; and vinyl chloride. Primary standards of these toxic compounds should be prepared in a chemical fume hood, and a NIOSH/MESA approved toxic gas respirator should be worn when handling high concentrations of these compounds.
4.3 This method allows the use of hydrogen as a carrier gas in place of helium (Section 5.3.1.2). The laboratory should take the necessary precautions in dealing with hydrogen, and should limit hydrogen flow at the source to prevent buildup of an explosive mixture of hydrogen in air.
5. Apparatus and Materials Note:Brand names, suppliers, and part numbers are cited for illustration purposes only. No endorsement is implied. Equivalent performance may be achieved using equipment and materials other than those specified here. Demonstration of equivalent performance that meets the requirements of this method is the responsibility of the laboratory. Suppliers for equipment and materials in this method may be found through an on-line search.
5.1 Sampling equipment for discrete sampling.
5.1.1 Vial - 25- or 40-mL capacity, or larger, with screw cap with a hole in the center (Fisher #13075 or equivalent). Unless pre-cleaned, detergent wash, rinse with tap and reagent water, and dry at 105 ± 5 °C before use.
5.1.2 Septum - Fluoropolymer-faced silicone (Fisher #12722 or equivalent). Unless pre-cleaned, detergent wash, rinse with tap and reagent water, and dry at 105 ± 5 °C for one hour before use.
5.2 Purge-and-trap system - The purge-and-trap system consists of three separate pieces of equipment: A purging device, trap, and desorber. Several complete systems are commercially available with autosamplers. Any system that meets the performance requirements in this method may be used.
5.2.1 The purging device should accept 5- to 25-mL samples with a water column at least 3 cm deep. The purge gas must pass though the water column as finely divided bubbles. The purge gas must be introduced no more than 5 mm from the base of the water column. Purge devices of a different volume may be used so long as the performance requirements in this method are met.
5.2.2 The trap should be at least 25 cm long and have an inside diameter of at least 0.105 in. The trap should be packed to contain the following minimum lengths of adsorbents: 1.0 cm of methyl silicone coated packing (section 6.3.2), 15 cm of 2,6-diphenylene oxide polymer (section 6.3.1), and 8 cm of silica gel (section 6.3.3). A trap with different dimensions and packing materials is acceptable so long as the performance requirements in this method are met.
5.2.3 The desorber should be capable of rapidly heating the trap to the temperature necessary to desorb the analytes of interest, and of maintaining this temperature during desorption. The trap should not be heated higher than the maximum temperature recommended by the manufacturer.
5.2.4 The purge-and-trap system may be assembled as a separate unit or coupled to a gas chromatograph.
5.3 GC/MS system.
5.3.1 Gas chromatograph (GC) - An analytical system complete with a temperature programmable gas chromatograph and all required accessories, including syringes and analytical columns. Autosamplers designed for purge-and-trap analysis of volatiles also may be used.
5.3.1.1 Injection port - Volatiles interface, split, splitless, temperature programmable split/splitless (PTV), large volume, on-column, backflushed, or other.
5.3.1.2 Carrier gas - Data in the tables in this method were obtained using helium carrier gas. If another carrier gas is used, analytical conditions may need to be adjusted for optimum performance, and calibration and all QC tests must be performed with the alternative carrier gas. See Section 4.3 for precautions regarding the use of hydrogen as a carrier gas.
5.3.2 GC column - See the footnote to Table 3. Other columns or column systems may be used provided all requirements in this method are met.
5.3.3 Mass spectrometer - Capable of repetitively scanning from 35-260 Daltons (amu) every 2 seconds or less, utilizing a 70 eV (nominal) electron energy in the electron impact ionization mode, and producing a mass spectrum which meets all criteria in Table 4 when 50 ng or less of 4-bromofluorobenzene (BFB) is injected through the GC inlet. If acrolein, acrylonitrile, chloromethane, and vinyl chloride are to be determined, it may be necessary to scan from below 25 Daltons to measure the peaks in the 26-35 Dalton range for reliable identification.
5.3.4 GC/MS interface - Any GC to MS interface that meets all performance requirements in this method may be used.
5.3.5 Data system - A computer system must be interfaced to the mass spectrometer that allows continuous acquisition and storage of mass spectra throughout the chromatographic program. The computer must have software that allows searching any GC/MS data file for specific m/z's (masses) and plotting m/z abundances versus time or scan number. This type of plot is defined as an extracted ion current profile (EICP). Software must also be available that allows integrating the abundance at any EICP between specified time or scan number limits.
5.4 Syringes - Graduated, 5-25 mL, glass hypodermic with Luerlok tip, compatible with the purging device.
5.5 Micro syringes - Graduated, 25-1000 µL, with 0.006 in. ID needle.
5.6 Syringe valve - Two-way, with Luer ends.
5.7 Syringe - 5 mL, gas-tight with shut-off valve.
5.8 Bottle - 15 mL, screw-cap, with Teflon cap liner.
5.9 Balance - Analytical, capable of accurately weighing 0.0001 g.
6. Reagents6.1 Reagent water - Reagent water is defined as water in which the analytes of interest and interfering compounds are not detected at the MDLs of the analytes of interest. It may be generated by passing deionized water, distilled water, or tap water through a carbon bed, passing the water through a water purifier, or heating the water to between 90 and 100 °C while bubbling contaminant-free gas through it for approximately 1 hour. While still hot, transfer the water to screw-cap bottles and seal with a fluoropolymer-lined cap.
6.2 Sodium thiosulfate - (ACS) Granular.
6.3 Trap materials.
6.3.1 2,6-Diphenylene oxide polymer - Tenax, 60/80 mesh, chromatographic grade, or equivalent.
6.3.2 Methyl silicone packing - 3% OV-1 on Chromosorb-W, 60/80 mesh, or equivalent.
6.3.3 Silica gel - 35/60 mesh, Davison, Grade-15 or equivalent.
6.3.4 Other trap materials are acceptable if performance requirements in this method are met.
6.4 Methanol - Demonstrated to be free from the target analytes and potentially interfering compounds.
6.5 Stock standard solutions - Stock standard solutions may be prepared from pure materials, or purchased as certified solutions. Traceability must be to the National Institute of Standards and Technology (NIST) or other national or international standard, when available. Stock solution concentrations alternative to those below may be used. Prepare stock standard solutions in methanol using assayed liquids or gases as appropriate. Because some of the compounds in this method are known to be toxic, primary dilutions should be prepared in a hood, and a NIOSH/MESA approved toxic gas respirator should be worn when high concentrations of neat materials are handled. The following procedure may be used to prepare standards from neat materials:
6.5.1 Place about 9.8 mL of methanol in a 10-mL ground-glass-stoppered volumetric flask. Allow the flask to stand, unstoppered, for about 10 minutes or until all alcohol wetted surfaces have dried. Weigh the flask to the nearest 0.1 mg.
6.5.2 Add the assayed reference material.
6.5.2.1 Liquids - Using a 100 µL syringe, immediately add two or more drops of assayed reference material to the flask. Be sure that the drops fall directly into the alcohol without contacting the neck of the flask. Reweigh, dilute to volume, stopper, then mix by inverting the flask several times. Calculate the concentration in µg/µL from the net gain in weight.
6.5.2.2 Gases - To prepare standards for any of compounds that boil below 30 °C, fill a 5-mL valved gas-tight syringe with reference standard vapor to the 5.0 mL mark. Lower the needle to 5 mm above the methanol meniscus. Slowly introduce the vapor above the surface of the liquid (the vapor will rapidly dissolve in the methanol). Reweigh, dilute to volume, stopper, then mix by inverting the flask several times. Calculate the concentration in µg/µL from the net gain in weight.
6.5.3 When compound purity is assayed to be 96% or greater, the weight may be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards may be used at any concentration if they are certified by the manufacturer or by an independent source.
6.5.4 Prepare fresh standards weekly for the gases and 2-chloroethylvinyl ether. Unless stated otherwise in this method, store non-aqueous standards in fluoropolymer-lined screw-cap, or heat-sealed, glass containers, in the dark at −20 to −10 °C. Store aqueous standards; e.g., the aqueous LCS (section 8.4.1) in the dark at ≤6 °C (but do not freeze) with zero headspace; e.g., in VOA vials (section 5.1.1). Standards prepared by the laboratory may be stored for up to one month, except when comparison with QC check standards indicates that a standard has degraded or become more concentrated due to evaporation, or unless the laboratory has data on file to prove stability for a longer period. Commercially prepared standards may be stored until the expiration date provided by the vendor, except when comparison with QC check standards indicates that a standard has degraded or become more concentrated due to evaporation, or unless the laboratory has data from the vendor on file to prove stability for a longer period.
Note:2-Chloroethylvinyl ether has been shown to be stable for as long as one month if prepared as a separate standard, and the other analytes have been shown to be stable for as long as 2 months if stored at less than −10 °C with minimal headspace in sealed, miniature inert-valved vials.
6.6 Secondary dilution standards - Using stock solutions, prepare secondary dilution standards in methanol that contain the compounds of interest, either singly or mixed. Secondary dilution standards should be prepared at concentrations such that the aqueous calibration standards prepared in section 7.3.2 will bracket the working range of the analytical system.
6.7 Surrogate standard spiking solution - Select a minimum of three surrogate compounds from Table 5. The surrogates selected should match the purging characteristics of the analytes of interest as closely as possible. Prepare a stock standard solution for each surrogate in methanol as described in section 6.5, and prepare a solution for spiking the surrogates into all blanks, LCSs, and MS/MSDs. Prepare the spiking solution such that spiking a small volume will result in a constant concentration of the surrogates. For example, add 10 µL of a spiking solution containing the surrogates at a concentration of 15 µg/mL in methanol to a 5-mL aliquot of water to produce a concentration of 30 µg/L for each surrogate. Other surrogate concentrations may be used. Store per section 6.5.4.
6.8 BFB standard - Prepare a solution of BFB in methanol as described in Sections 6.5 and 6.6. The solution should be prepared such that an injection or purging from water will result in introduction of ≤ 50 ng into the GC. BFB may be included in a mixture with the internal standards and/or surrogates.
6.9 Quality control check sample concentrate - See Section 8.2.1.
7. Calibration7.1 Assemble a purge-and-trap system that meets the specifications in Section 5.2. Prior to first use, condition the trap overnight at 180 °C by backflushing with gas at a flow rate of at least 20 mL/min. Condition the trap after each analysis at a temperature and time sufficient to prevent detectable concentrations of the analytes or contaminants in successive analyses.
7.2 Connect the purge-and-trap system to the gas chromatograph. The gas chromatograph should be operated using temperature and flow rate conditions equivalent to those given in the footnotes to Table 3. Alternative temperature and flow rate conditions may be used provided that performance requirements in this method are met.
7.3 Internal standard calibration.
7.3.1 Internal standards.
7.3.1.1 Select three or more internal standards similar in chromatographic behavior to the compounds of interest. Suggested internal standards are listed in Table 5. Use the base peak m/z as the primary m/z for quantification of the standards. If interferences are found at the base peak, use one of the next two most intense m/z's for quantitation. Demonstrate that measurements of the internal standards are not affected by method or matrix interferences.
7.3.1.2 To assure accurate analyte identification, particularly when selected ion monitoring (SIM) is used, it may be advantageous to include more internal standards than those suggested in Section 7.3.1.1. An analyte will be located most accurately if its retention time relative to an internal standard is in the range of 0.8 to 1.2.
7.3.1.3 Prepare a stock standard solution for each internal standard in methanol as described in Section 6.5, and prepare a solution for spiking the internal standards into all blanks, LCSs, and MS/MSDs. Prepare the spiking solution such that spiking a small volume will result in a constant concentration of the internal standards. For example, add 10 µL of a spiking solution containing the internal standards at a concentration of 15 µg/mL in methanol to a 5-mL aliquot of water to produce a concentration of 30 µg/L for each internal standard. Other concentrations may be used. The internal standard solution and the surrogate standard spiking solution (Section 6.7) may be combined, if desired. Store per section 6.5.4.
7.3.2 Calibration.
7.3.2.1 Calibration standards.
7.3.2.1.1 Prepare calibration standards at a minimum of five concentration levels for each analyte of interest by adding appropriate volumes of one or more stock standards to a fixed volume (e.g., 40 mL) of reagent water in volumetric glassware. Fewer levels may be necessary for some analytes based on the sensitivity of the MS, but no fewer than 3 levels may be used, and only the highest or lowest point(s) may be dropped from the calibration. One of the calibration standards should be at a concentration at or below the ML or as specified by a regulatory/control authority or in a permit. The ML value may be rounded to a whole number that is more convenient for preparing the standard, but must not exceed the ML values listed in Table 1 for those analytes which list ML values. Alternatively, the laboratory may establish the ML for each analyte based on the concentration of the lowest calibration standard in a series of standards produced in the laboratory or obtained from a commercial vendor, again, provided that the ML value does not exceed the MLs in Table 1, and provided that the resulting calibration meets the acceptance criteria in Section 7.3.4, based on the RSD, RSE, or R 2. The concentrations of the higher standards should correspond to the expected range of concentrations found in real samples, or should define the working range of the GC/MS system for full-scan and/or SIM operation, as appropriate. A minimum of six concentration levels is required for a second order, non-linear (e.g., quadratic; ax 2 + bx + c = 0) calibration. Calibrations higher than second order are not allowed.
7.3.2.1.2 To each calibration standard or standard mixture, add a known constant volume of the internal standard spiking solution (section 7.3.1.3) and surrogate standard spiking solution (section 6.7) or the combined internal standard solution and surrogate spiking solution (section 7.3.1.3). Aqueous standards may be stored up to 24 hours, if held in sealed vials with zero headspace. If not so stored, they must be discarded after one hour.
7.3.2.2 Prior to analysis of the calibration standards, analyze the BFB standard (section 6.8) and adjust the scan rate of the MS to produce a minimum of 5 mass spectra across the BFB GC peak, but do not exceed 2 seconds per scan. Adjust instrument conditions until the BFB criteria in Table 4 are met. Once the scan conditions are established, they must be used for analyses of all standards, blanks, and samples.
Note:The BFB spectrum may be evaluated by summing the intensities of the m/z's across the GC peak, subtracting the background at each m/z in a region of the chromatogram within 20 scans of but not including any part of the BFB peak. The BFB spectrum may also be evaluated by fitting a Gaussian to each m/z and using the intensity at the maximum for each Gaussian, or by integrating the area at each m/z and using the integrated areas. Other means may be used for evaluation of the BFB spectrum so long as the spectrum is not distorted to meet the criteria in Table 4.
7.3.2.3 Analyze the mid-point standard and enter or review the retention time, relative retention time, mass spectrum, and quantitation m/z in the data system for each analyte of interest, surrogate, and internal standard. If additional analytes (Table 2) are to be quantified, include these analytes in the standard. The mass spectrum for each analyte must be comprised of a minimum of 2 m/z's; 3 to 5 m/z's assure more reliable analyte identification. Suggested quantitation m/z's are shown in Table 6 as the primary m/z. For analytes in Table 6 that do not have a secondary m/z, acquire a mass spectrum and enter one or more secondary m/z's for more reliable identification. If an interference occurs at the primary m/z, use one of the secondary m/z's or an alternative m/z. A single m/z only is required for quantitation.
7.3.2.4 For SIM operation, determine the analytes in each descriptor, the quantitation m/z for each analyte (the quantitation m/z can be the same as for full-scan operation; Section 7.3.2.3), the dwell time on each m/z for each analyte, and the beginning and ending retention time for each descriptor. Analyze the verification standard in scan mode to verify m/z's and establish retention times for the analytes. There must be a minimum of two m/z's for each analyte to assure analyte identification. To maintain sensitivity, the number of m/z's in a descriptor should be limited. For example, for a descriptor with 10 m/z's and a chromatographic peak width of 5 sec, a dwell time of 100 ms at each m/z would result in a scan time of 1 second and provide 5 scans across the GC peak. The quantitation m/z will usually be the most intense peak in the mass spectrum. The quantitation m/z and dwell time may be optimized for each analyte. The acquisition table used for SIM must take into account the mass defect (usually less than 0.2 Dalton) that can occur at each m/z monitored. Refer to the footnotes to Table 3 for establishing operating conditions and to section 7.3.2.2 for establishing scan conditions.
7.3.2.5 For combined scan and SIM operation, set up the scan segments and descriptors to meet requirements in sections 7.3.2.2-7.3.2.4. Analyze unfamiliar samples in the scan mode to assure that the analytes of interest are determined.
7.3.3 Analyze each calibration standard according to Section 10 and tabulate the area at the quantitation m/z against concentration for each analyte of interest, surrogate, and internal standard. Calculate the response factor (RF) for each compound at each concentration using Equation 1.
 Where: As
= Area of the characteristic m/z for the analyte to be measured.
Ais = Area of the characteristic m/z for the internal standard. Cis
= Concentration of the internal standard (µg/L). Cs = Concentration
of the analyte to be measured (µg/L).
Where: As
= Area of the characteristic m/z for the analyte to be measured.
Ais = Area of the characteristic m/z for the internal standard. Cis
= Concentration of the internal standard (µg/L). Cs = Concentration
of the analyte to be measured (µg/L).
7.3.4 Calculate the mean (average) and relative standard deviation (RSD) of the response factors. If the RSD is less than 35%, the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to fit a linear or quadratic regression of response ratios, As/Ais, vs. concentration ratios Cs/Cis. If used, the regression must be weighted inversely proportional to concentration (1/C). The coefficient of determination (R 2) of the weighted regression must be greater than 0.920 (this value roughly corresponds to the RSD limit of 35%). Alternatively, the relative standard error (Reference 10) may be used as an acceptance criterion. As with the RSD, the RSE must be less than 35%. If an RSE less than 35% cannot be achieved for a quadratic regression, system performance is unacceptable, and the system must be adjusted and re-calibrated.
Note:Using capillary columns and current instrumentation, it is quite likely that a laboratory can calibrate the target analytes in this method and achieve a linearity metric (either RSD or RSE) well below 35%. Therefore, laboratories are permitted to use more stringent acceptance criteria for calibration than described here, for example, to harmonize their application of this method with those from other sources.
7.4 Calibration verification - Because the analytical system is calibrated by purge of the analytes from water, calibration verification is performed using the laboratory control sample (LCS). See section 8.4 for requirements for calibration verification using the LCS, and the Glossary for further definition.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality assurance program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and ongoing analysis of spiked samples and blanks to evaluate and document data quality (40 CFR 136.7). The laboratory must maintain records to document the quality of data generated. Results of ongoing performance tests are compared with established QC acceptance criteria to determine if the results of analyses meet performance requirements of this method. When results of spiked samples do not meet the QC acceptance criteria in this method, a quality control check sample (laboratory control sample; LCS) must be analyzed to confirm that the measurements were performed in an in-control mode of operation. A laboratory may develop its own performance criteria (as QC acceptance criteria), provided such criteria are as or more restrictive than the criteria in this method.
8.1.1 The laboratory must make an initial demonstration of capability (DOC) to generate acceptable precision and recovery with this method. This demonstration is detailed in Section 8.2. On a continuing basis, the laboratory must repeat demonstration of capability (DOC) at least annually.
8.1.2 In recognition of advances that are occurring in analytical technology, and to overcome matrix interferences, the laboratory is permitted certain options (section 1.5 and 40 CFR 136.6(b)) to improve separations or lower the costs of measurements. These options may include an alternative purge-and-trap device, and changes in both column and type of mass spectrometer (see 40 CFR 136.6(b)(4)(xvi)). Alternative determinative techniques, such as substitution of spectroscopic or immunoassay techniques, and changes that degrade method performance, are not allowed. If an analytical technique other than GC/MS is used, that technique must have a specificity equal to or greater than the specificity of GC/MS for the analytes of interest. The laboratory is also encouraged to participate in inter-comparison and performance evaluation studies (see section 8.8).
8.1.2.1 Each time a modification is made to this method, the laboratory is required to repeat the procedure in section 8.2. If the detection limit of the method will be affected by the change, the laboratory must demonstrate that the MDLs (40 CFR part 136, appendix B) are lower than one-third the regulatory compliance limit or the MDLs in this method, whichever are greater. If calibration will be affected by the change, the instrument must be recalibrated per section 7. Once the modification is demonstrated to produce results equivalent or superior to results produced by this method, that modification may be used routinely thereafter, so long as the other requirements in this method are met (e.g., matrix spike/matrix spike duplicate recovery and relative percent difference).
8.1.2.1.1 If a modification is to be applied to a specific discharge, the laboratory must prepare and analyze matrix spike/matrix spike duplicate (MS/MSD) samples (Section 8.3) and LCS samples (section 8.4). The laboratory must include internal standards and surrogates (section 8.7) in each of the samples. The MS/MSD and LCS samples must be fortified with the analytes of interest (section 1.3.). If the modification is for nationwide use, MS/MSD samples must be prepared from a minimum of nine different discharges (See section 8.1.2.1.2), and all QC acceptance criteria in this method must be met. This evaluation only needs to be performed once, other than for the routine QC required by this method (for example it could be performed by the vendor of the alternative materials) but any laboratory using that specific material must have the results of the study available. This includes a full data package with the raw data that will allow an independent reviewer to verify each determination and calculation performed by the laboratory (see section 8.1.2.2.5, items (a)-(l)).
8.1.2.1.2 Sample matrices on which MS/MSD tests must be performed for nationwide use of an allowed modification:
(a) Effluent from a publicly owned treatment works (POTW).
(b) ASTM D5905 Standard Specification for Substitute Wastewater.
(c) Sewage sludge, if sewage sludge will be in the permit.
(d) ASTM D1141 Standard Specification for Substitute Ocean Water, if ocean water will be in the permit.
(e) Untreated and treated wastewaters up to a total of nine matrix types (see https://www.epa.gov/eg/industrial-effluent-guidelines for a list of industrial categories with existing effluent guidelines).
(i) At least one of the above wastewater matrix types must have at least one of the following characteristics:
(A) Total suspended solids greater than 40 mg/L.
(B) Total dissolved solids greater than 100 mg/L.
(C) Oil and grease greater than 20 mg/L.
(D) NaCl greater than 120 mg/L.
(E) CaCO3 greater than 140 mg/L.
(ii) Results of MS/MSD tests must meet QC acceptance criteria in section 8.3.
(f) A proficiency testing (PT) sample from a recognized provider, in addition to tests of the nine matrices (section 8.1.2.1.1).
8.1.2.2 The laboratory is required to maintain records of modifications made to this method. These records include the following, at a minimum:
8.1.2.2.1 The names, titles, and business street addresses, telephone numbers, and email addresses of the analyst(s) that performed the analyses and modification, and of the quality control officer that witnessed and will verify the analyses and modifications.
8.1.2.2.2 A list of analytes, by name and CAS Registry Number.
8.1.2.2.3 A narrative stating reason(s) for the modifications.
8.1.2.2.4 Results from all quality control (QC) tests comparing the modified method to this method, including:
(a) Calibration (section 7).
(b) Calibration verification/LCS (section 8.4).
(c) Initial demonstration of capability (section 8.2).
(d) Analysis of blanks (section 8.5).
(e) Matrix spike/matrix spike duplicate analysis (section 8.3).
(f) Laboratory control sample analysis (section 8.4).
8.1.2.2.5 Data that will allow an independent reviewer to validate each determination by tracing the instrument output (peak height, area, or other signal) to the final result. These data are to include:
(a) Sample numbers and other identifiers.
(b) Analysis dates and times.
(c) Analysis sequence/run chronology.
(d) Sample volume (Section 10).
(e) Sample dilution (Section 13.2).
(f) Instrument and operating conditions.
(g) Column (dimensions, material, etc).
(h) Operating conditions (temperature program, flow rate, etc).
(i) Detector (type, operating conditions, etc).
(j) Chromatograms, mass spectra, and other recordings of raw data.
(k) Quantitation reports, data system outputs, and other data to link the raw data to the results reported.
(l) A written Standard Operating Procedure (SOP).
8.1.2.2.6 Each individual laboratory wishing to use a given modification must perform the start-up tests in section 8.1.2 (e.g., DOC, MDL), with the modification as an integral part of this method prior to applying the modification to specific discharges. Results of the DOC must meet the QC acceptance criteria in Table 7 for the analytes of interest (section 1.3), and the MDLs must be equal to or lower than the MDLs in Table3 for the analytes of interest
8.1.3 Before analyzing samples, the laboratory must analyze a blank to demonstrate that interferences from the analytical system, labware, and reagents are under control. Each time a batch of samples is analyzed or reagents are changed, a blank must be analyzed as a safeguard against laboratory contamination. Requirements for the blank are given in section 8.5.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze samples to monitor and evaluate method and laboratory performance on the sample matrix. The procedure for spiking and analysis is given in section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through analysis of a quality control check sample (laboratory control sample, LCS; on-going precision and recovery sample, OPR) that the measurement system is in control. This procedure is given in section 8.4.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is given in section 8.8.
8.1.7 The large number of analytes tested in performance tests in this method present a substantial probability that one or more will fail acceptance criteria when many analytes are tested simultaneously, and a re-test is allowed if this situation should occur. If, however, continued re-testing results in further repeated failures, the laboratory must document and report the failures (e.g., as qualifiers on results), unless the failures are not required to be reported as determined by the regulatory/control authority. Results associated with a QC failure for an analyte regulated in a discharge cannot be used to demonstrate regulatory compliance. QC failures do not relieve a discharger or permittee of reporting timely results.
8.2 Initial demonstration of capability (DOC) - To establish the ability to generate acceptable recovery and precision, the laboratory must perform the DOC in sections 8.2.1 through 8.2.6 for the analytes of interest. The laboratory must also establish MDLs for the analytes of interest using the MDL procedure at 40 CFR part 136, appendix B. The laboratory's MDLs must be equal to or lower than those listed in Table 1 for those analytes which list MDL values, or lower than one-third the regulatory compliance limit, whichever is greater. For MDLs not listed in Table 1, the laboratory must determine the MDLs using the MDL procedure at 40 CFR part 136, appendix B under the same conditions used to determine the MDLs for the analytes listed in Table 1. All procedures used in the analysis must be included in the DOC.
8.2.1 For the DOC, a QC check sample concentrate (LCS concentrate) containing each analyte of interest (section 1.3) is prepared in methanol. The QC check sample concentrate must be prepared independently from those used for calibration, but may be from the same source as the second-source standard used for calibration verification/LCS (sections 7.4 and 8.4). The concentrate should produce concentrations of the analytes of interest in water at the mid-point of the calibration range, and may be at the same concentration as the LCS (section 8.4).
Note:QC check sample concentrates are no longer available from EPA.
8.2.2 Using a pipet or micro-syringe, prepare four LCSs by adding an appropriate volume of the concentrate to each of four aliquots of reagent water. The volume of reagent water must be the same as the volume that will be used for the sample, blank (section 8.5), and MS/MSD (section 8.3). A volume of 5 mL and a concentration of 20 µg/L were used to develop the QC acceptance criteria in Table 7. An alternative volume and sample concentration may be used, provided that all QC tests are performed and all QC acceptance criteria in this method are met. Also add an aliquot of the surrogate spiking solution (section 6.7) and internal standard spiking solution (section 7.3.1.3) to the reagent-water aliquots.
8.2.3 Analyze the four LCSs according to the method beginning in section 10.
8.2.4 Calculate the average percent recovery (X) and the standard deviation of the percent recovery (s) for each analyte using the four results.
8.2.5 For each analyte, compare s and X with the corresponding acceptance criteria for precision and recovery in Table 7. For analytes in Tables 1 and 2 not listed in Table 7, DOC QC acceptance criteria must be developed by the laboratory. EPA has provided guidance for development of QC acceptance criteria (References 11 and 12). Alternatively, acceptance criteria for analytes not listed in Table 7 may be based on laboratory control charts. If s and X for all analytes of interest meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may begin. If any individual s exceeds the precision limit or any individual X falls outside the range for recovery, system performance is unacceptable for that analyte.
Note:The large number of analytes in Tables 1 and 2 present a substantial probability that one or more will fail at least one of the acceptance criteria when many or all analytes are determined simultaneously. Therefore, the analyst is permitted to conduct a “re-test” as described in section 8.2.6.
8.2.6 When one or more of the analytes tested fail at least one of the acceptance criteria, repeat the test for only the analytes that failed. If results for these analytes pass, system performance is acceptable and analysis of samples and blanks may proceed. If one or more of the analytes again fail, system performance is unacceptable for the analytes that failed the acceptance criteria. Correct the problem and repeat the test (section 8.2). See section 8.1.7 for disposition of repeated failures.
Note:To maintain the validity of the test and re-test, system maintenance and/or adjustment is not permitted between this pair of tests.
8.3 Matrix spike and matrix spike duplicate (MS/MSD) - The purpose of the MS/MSD requirement is to provide data that demonstrate the effectiveness of the method as applied to the samples in question by a given laboratory, and both the data user (discharger, permittee, regulated entity, regulatory/control authority, customer, other) and the laboratory share responsibility for provision of such data. The data user should identify the sample and the analytes of interest (section 1.3) to be spiked and provide sufficient sample volume to perform MS/MSD analyses. The laboratory must, on an ongoing basis, spike at least 5% of the samples in duplicate from each discharge being monitored to assess accuracy (recovery and precision). If direction cannot be obtained from the data user, the laboratory must spike at least one sample in duplicate per extraction batch of up to 20 samples with the analytes in Table 1. Spiked sample results should be reported only to the data user whose sample was spiked, or as requested or required by a regulatory/control authority, or in a permit.
8.3.1 If, as in compliance monitoring, the concentration of a specific analyte will be checked against a regulatory concentration limit, the concentration of the spike should be at that limit; otherwise, the concentration of the spike should be one to five times higher than the background concentration determined in section 8.3.2, at or near the mid-point of the calibration range, or at the concentration in the LCS (section 8.4) whichever concentration would be larger.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of the each analyte of interest. If necessary, prepare a new check sample concentrate (section 8.2.1) appropriate for the background concentration. Spike and analyze two additional sample aliquots, and determine the concentration after spiking (A1 and A2) of each analyte. Calculate the percent recoveries (P1 and P2) as 100 (A1−B)/T and 100 (A2−B)/T, where T is the known true value of the spike. Also calculate the relative percent difference (RPD) between the concentrations (A1 and A2) as 200 |A1−A2|/(A1 + A2). If necessary, adjust the concentrations used to calculate the RPD to account for differences in the volumes of the spiked aliquots.
8.3.3 Compare the percent recoveries (P1 and P2) and the RPD for each analyte in the MS/MSD aliquots with the corresponding QC acceptance criteria in Table 7. A laboratory may develop and apply QC acceptance criteria more restrictive than the criteria in Table 7, if desired.
8.3.3.1 If any individual P falls outside the designated range for recovery in either aliquot, or the RPD limit is exceeded, the result for the analyte in the unspiked sample is suspect. See Section 8.1.7 for disposition of failures.
8.3.3.2 The acceptance criteria in Table 7 were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the spike to background ratio approaches 5:1 (Reference 13) and is applied to spike concentrations of 20 µg/L and higher. If spiking is performed at a concentration lower than 20 µg/L, the laboratory must use the QC acceptance criteria in Table 7, the optional QC acceptance criteria calculated for the specific spike concentration in Table 8, or optional in-house criteria (Section 8.3.4). To use the acceptance criteria in Table 8: (1) Calculate recovery (X') using the equation in Table 8, substituting the spike concentration (T) for C; (2) Calculate overall precision (S') using the equation in Table 8, substituting X' for X; (3) Calculate the range for recovery at the spike concentration as (100 X'/T) ± 2.44(100 S'/T)% (Reference 4). For analytes of interest in Tables 1 and 2 not listed in Table 7, QC acceptance criteria must be developed by the laboratory. EPA has provided guidance for development of QC acceptance criteria (References 11 and 12). Alternatively, acceptance criteria may be based on laboratory control charts. In-house LCS QC acceptance criteria must be updated at least every two years.
8.3.4 After analysis of a minimum of 20 MS/MSD samples for each target analyte and surrogate, and if the laboratory chooses to develop and apply in-house QC limits, the laboratory should calculate and apply in-house QC limits for recovery and RPD of future MS/MSD samples (section 8.3). The QC limits for recovery are calculated as the mean observed recovery ± 3 standard deviations, and the upper QC limit for RPD is calculated as the mean RPD plus 3 standard deviations of the RPDs. The in-house QC limits must be updated at least every two years and re-established after any major change in the analytical instrumentation or process. If in-house QC limits are developed, at least 80% of the analytes tested in the MS/MSD must have in-house QC acceptance criteria that are tighter than those in Table 7 and the remaining analytes (those other than the analytes included in the 80%) must meet the acceptance criteria in Table 7. If an in-house QC limit for the RPD is greater than the limit in Table 7, then the limit in Table 7 must be used. Similarly, if an in-house lower limit for recovery is below the lower limit in Table 7, then the lower limit in Table 7 must be used, and if an in-house upper limit for recovery is above the upper limit in Table 7, then the upper limit in Table 7 must be used.
8.4 Calibration verification/laboratory control sample (LCS) - The working calibration curve or RF must be verified immediately after calibration and at the beginning of each 12-hour shift by the measurement of an LCS. The LCS must be from a source different from the source used for calibration (section 7.3.2.1), but may be the same as the sample prepared for the DOC (section 8.2.1).
Note:The 12-hour shift begins after analysis of BFB, the LCS, and the blank, and ends 12 hours later. BFB, the LCS, and blank are outside of the 12-hour shift (Section 11.4). The MS and MSD are treated as samples and are analyzed within the 12-hour shift.
8.4.1 Prepare the LCS by adding QC check sample concentrate (section 8.2.1) to reagent water. Include all analytes of interest (Section 1.3) in the LCS. The volume of reagent water must be the same as the volume used for the sample, blank (Section 8.5), and MS/MSD (section 8.3). Also add an aliquot of the surrogate solution (Section 6.7) and internal standard solution (section 7.3.1.3). The concentration of the analytes in reagent water should be the same as the concentration in the DOC (section 8.2.2).
8.4.2 Analyze the LCS prior to analysis of field samples in the batch of samples analyzed during the 12-hour shift (see the Note at section 8.4). Determine the concentration (A) of each analyte. Calculate the percent recovery (Q) as 100 (A/T) %, where T is the true value of the concentration in the LCS.
8.4.3 Compare the percent recovery (Q) for each analyte with its corresponding QC acceptance criterion in Table 7. For analytes of interest in Tables 1 and 2 not listed in Table 7, use the QC acceptance criteria developed for the LCS (section 8.4.5). If the recoveries for all analytes of interest fall within their respective QC acceptance criteria, analysis of blanks and field samples may proceed. If any individual Q falls outside the range, proceed according to section 8.4.4.
Note:The large number of analytes in Tables 1 - 2 present a substantial probability that one or more will fail the acceptance criteria when all analytes are tested simultaneously. Because a re-test is allowed in event of failure (sections 8.1.7 and 8.4.3), it may be prudent to analyze two LCSs together and evaluate results of the second analysis against the QC acceptance criteria only if an analyte fails the first test.
8.4.4 Repeat the test only for those analytes that failed to meet the acceptance criteria (Q). If these analytes now pass, system performance is acceptable and analysis of blanks and samples may proceed. Repeated failure, however, will confirm a general problem with the measurement system. If this occurs, repeat the test (section 8.4.2). using a fresh LCS (section 8.2.2) or an LCS prepared with a fresh QC check sample concentrate (section 8.2.1), or perform and document system repair. Subsequent to repair, repeat the calibration verification/LCS test (section 8.4). If the acceptance criteria for Q cannot be met, re-calibrate the instrument (section 7). See section 8.1.7 for disposition of repeated failures.
Note:To maintain the validity of the test and re-test, system maintenance and/or adjustment is not permitted between the pair of tests.
8.4.5 After analysis of 20 LCS samples, and if the laboratory chooses to develop and apply in-house QC limits, the laboratory should calculate and apply in-house QC limits for recovery to future LCS samples (section 8.4). Limits for recovery in the LCS calculated as the mean recovery ±3 standard deviations. A minimum of 80% of the analytes tested for in the LCS must have QC acceptance criteria tighter than those in Table 7, and the remaining analytes (those other than the analytes included in the 80%) must meet the acceptance criteria in Table 7. If an in-house lower limit for recovery is lower than the lower limit in Table 7, the lower limit in Table 7 must be used, and if an in-house upper limit for recovery is higher than the upper limit in Table 7, the upper limit in Table 7 must be used. Many of the analytes and surrogates do not have acceptance criteria. The laboratory should use 60-140% as interim acceptance criteria for recoveries of spiked analytes that do not have recovery limits specified in Table 7, and least 80% of the analytes should meet the 60-140% interim criteria until in-house LCS limits are developed. Alternatively, acceptance criteria for analytes that do not have recovery limits in Table 7 may be based on laboratory control charts. In-house QC acceptance criteria must be updated at least every two years.
8.5 Blank - A blank must be analyzed prior to each 12-hour shift to demonstrate freedom from contamination. A blank must also be analyzed after a sample containing a high concentration of an analyte or potentially interfering compound to demonstrate freedom from carry-over.
8.5.1 Spike the internal standards and surrogates into the blank. Analyze the blank immediately after analysis of the LCS (Section 8.4) and prior to analysis of the MS/MSD and samples to demonstrate freedom from contamination.
8.5.2 If any analyte of interest is found in the blank: At a concentration greater than the MDL for the analyte, at a concentration greater than one-third the regulatory compliance limit, or at a concentration greater than one-tenth the concentration in a sample analyzed during the 12-hour shift (section 8.4), whichever is greater; analysis of samples must be halted and samples affected by the blank must be re-analyzed. If, however, continued re-testing results in repeated blank contamination, the laboratory must document and report the failures (e.g., as qualifiers on results), unless the failures are not required to be reported as determined by the regulatory/control authority. Results associated with blank contamination for an analyte regulated in a discharge cannot be used to demonstrate regulatory compliance. QC failures do not relieve a discharger or permittee of reporting timely results.
8.6 Surrogate recoveries - The laboratory must evaluate surrogate recovery data in each sample against its in-house surrogate recovery limits for surrogates that do not have acceptance criteria in Table 7. The laboratory may use 60-140% as interim acceptance criteria for recoveries for surrogates not listed in Table 5. At least 80% of the surrogates must meet the 60-140% interim criteria until in-house limits are developed. Alternatively, surrogate recovery limits may be developed from laboratory control charts.
8.6.1 Spike the surrogates into all samples, blanks, LCSs, and MS/MSDs. Compare surrogate recoveries against the QC acceptance criteria in Table 7. For surrogates in Table 5 without QC acceptance criteria in Table 7, and for other surrogates that may be used by the laboratory, limits must be developed by the laboratory. EPA has provided guidance for development of QC acceptance criteria (References 11 and 12). Alternatively, surrogate recovery limits may be developed from laboratory control charts. In-house QC acceptance criteria must be updated at least every two years.
8.6.2 If any recovery fails its criteria, attempt to find and correct the cause of the failure. See section 8.1.7 for disposition of failures.
8.7 Internal standard responses.
8.7.1 Calibration verification/LCS - The responses (GC peak heights or areas) of the internal standards in the calibration verification/LCS must be within 50% to 200% (1/2 to 2×) of their respective responses in the mid-point calibration standard. If they are not, repeat the LCS test using a fresh QC check sample (section 8.4.1) or perform and document system repair. Subsequent to repair, repeat the calibration verification/LCS test (section 8.4). If the responses are still not within 50% to 200%, re-calibrate the instrument (section 7) and repeat the calibration verification/LCS test.
8.7.2 Samples, blanks, and MS/MSDs - The responses (GC peak heights or areas) of each internal standard in each sample, blank, and MS/MSD must be within 50% to 200% (1/2 to 2×) of its respective response in the mid-point calibration standard. If, as a group, all internal standards are not within this range, perform and document system repair, repeat the calibration verification/LCS test (section 8.4), and re-analyze the affected samples. If a single internal standard is not within the 50% to 200% range, use an alternative internal standard for quantitation of the analyte referenced to the affected internal standard. It may be necessary to use the data system to calculate a new response factor from calibration data for the alternative internal standard/analyte pair. If an internal standard fails the 50-200% criteria and no analytes are detected in the sample, ignore the failure or report it if required by the regulatory/control authority.
8.8 As part of the QC program for the laboratory, control charts or statements of accuracy for wastewater samples must be assessed and records maintained periodically (see 40 CFR 136.7(c)(1)(viii)). After analysis of five or more spiked wastewater samples as in section 8.3, calculate the average percent recovery (PX) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent interval from PX−2sp to PX + 2sp. For example, if PX = 90% and sp = 10%, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each analyte on a regular basis (e.g., after each 5-10 new accuracy measurements). If desired, statements of accuracy for laboratory performance, independent of performance on samples, may be developed using LCSs.
8.9 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of environmental measurements. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Collect the sample as a grab sample in a glass container having a total volume of at least 25 mL. Fill the sample bottle just to overflowing in such a manner that no air bubbles pass through the sample as the bottle is being filled. Seal the bottle so that no air bubbles are entrapped in it. If needed, collect additional sample(s) for the MS/MSD (section 8.3).
9.2 Ice or refrigerate samples at ≤6 °C from the time of collection until analysis, but do not freeze. If residual chlorine is present, add sodium thiosulfate preservative (10 mg/40 mL is sufficient for up to 5 ppm Cl2) to the empty sample bottle just prior to shipping to the sampling site. Any method suitable for field use may be employed to test for residual chlorine (Reference 14). Field test kits are also available for this purpose. If sodium thiosulfate interferes in the determination of the analytes, an alternative preservative (e.g., ascorbic acid or sodium sulfite) may be used. If preservative has been added, shake the sample vigorously for one minute. Maintain the hermetic seal on the sample bottle until time of analysis.
9.3 If acrolein is to be determined, analyze the sample within 3 days. To extend the holding time to 14 days, acidify a separate sample to pH 4-5 with HCl using the procedure in section 9.7.
9.4 Experimental evidence indicates that some aromatic compounds, notably benzene, toluene, and ethyl benzene are susceptible to rapid biological degradation under certain environmental conditions (Reference 3). Refrigeration alone may not be adequate to preserve these compounds in wastewaters for more than seven days. To extend the holding time for aromatic compounds to 14 days, acidify the sample to approximately pH 2 using the procedure in section 9.7.
9.5 If halocarbons are to be determined, either use the acidified aromatics sample in section 9.4 or acidify a separate sample to a pH of about 2 using the procedure in section 9.7.
9.6 The ethers listed in Table 2 are prone to hydrolysis at pH 2 when a heated purge is used. Aqueous samples should not be acid preserved if these ethers are of interest, or if the alcohols they would form upon hydrolysis are of interest and the ethers are anticipated to present.
9.7 Sample acidification - Collect about 500 mL of sample in a clean container and adjust the pH of the sample to 4-5 for acrolein (section 9.3), or to about 2 for the aromatic compounds (section 9.4) by adding 1+1 HCl while swirling or stirring. Check the pH with narrow range pH paper. Fill a sample container as described in section 9.1. Alternatively, fill a precleaned vial (section 5.1.1) that contains approximately 0.25 mL of 1+1 HCl with sample as in section 9.1. If preserved using this alternative procedure, the pH of the sample can be verified to be <2 after some of the sample is removed for analysis. Acidification will destroy 2-chloroethylvinyl ether; therefore, determine 2-chloroethylvinyl ether from the unacidified sample.
9.8 All samples must be analyzed within 14 days of collection (Reference 3), unless specified otherwise in sections 9.3-9.7.
10. Sample Purging and Gas Chromatography10.1 The footnote to Table 3 gives the suggested GC column and operating conditions MDLs and MLs for many of the analytes are given in Table 1. Retention times for many of the analytes are given in Table 3. Sections 10.2 through 10.7 suggest procedures that may be used with a manual purge-and-trap system. Auto-samplers and other columns or chromatographic conditions may be used if requirements in this method are met. Prior to performing analyses, and between analyses, it may be necessary to bake the purge-and-trap and GC systems (section 3.3).
10.2 Attach the trap inlet to the purging device, and set the purge-and-trap system to purge. Open the syringe valve located on the purging device sample introduction needle.
10.3 Allow the sample to come to ambient temperature prior to pouring an aliquot into the syringe. Remove the plunger from a syringe and attach a closed syringe valve. Open the sample bottle (or standard) and carefully pour the sample into the syringe barrel to just short of overflowing. Replace the syringe plunger and compress the sample. Open the syringe valve and vent any residual air while adjusting the sample volume. Since this process of taking an aliquot destroys the validity of the sample for future analysis, the analyst should fill a second syringe at this time to protect against possible loss of data. Add the surrogate spiking solution (section 6.7) and internal standard spiking solution (section 7.3.1.3) through the valve bore, then close the valve. The surrogate and internal standards may be mixed and added as a single spiking solution. Autosamplers designed for purge-and-trap analysis of volatiles also may be used.
10.4 Attach the syringe valve assembly to the syringe valve on the purging device. Open the syringe valve and inject the sample into the purging chamber.
10.5 Close both valves and purge the sample at a temperature, flow rate, and duration sufficient to purge the less-volatile analytes onto the trap, yet short enough to prevent blowing the more-volatile analytes through the trap. The temperature, flow rate, and time should be determined by test. The same purge temperature, flow rate, and purge time must be used for all calibration, QC, and field samples.
10.6 After the purge, set the purge-and-trap system to the desorb mode, and begin the temperature program of the gas chromatograph. Introduce the trapped materials to the GC column by rapidly heating the trap to the desorb temperature while backflushing the trap with carrier gas at the flow rate and for the time necessary to desorb the analytes of interest. The optimum temperature, flow rate, and time should be determined by test. The same temperature, desorb time, and flow rate must be used for all calibration, QC, and field samples. If heating of the trap does not result in sharp peaks for the early eluting analytes, the GC column may be used as a secondary trap by cooling to an ambient or subambient temperature. To avoid carry-over and interferences, maintain the trap at the desorb temperature and flow rate until the analytes, interfering compounds, and excess water are desorbed. The optimum conditions should be determined by test.
10.7 Start MS data acquisition at the start of the desorb cycle and stop data collection when the analytes of interest, potentially interfering compounds, and water have eluted (see the footnote to Table 3 for conditions).
10.8 Cool the trap to the purge temperature and return the trap to the purge mode. When the trap is cool, the next sample can be analyzed.
11. Performance Tests11.1 At the beginning of each 12-hour shift during which standards or samples will be analyzed, perform the tests in sections 11.2-11.3 to verify system performance. Use the instrument operating conditions in the footnotes to Table 3 for these performance tests. Alternative conditions may be used so as long as all QC requirements are met.
11.2 BFB - Inject 50 ng of BFB solution directly on the column. Alternatively, add BFB to reagent water or an aqueous standard such that 50 ng or less of BFB will be introduced into the GC. Analyze according to section 10. Confirm that all criteria in section 7.3.2.2 and Table 4 are met. If all criteria are not met, perform system repair, retune the mass spectrometer, and repeat the test until all criteria are met.
11.3 Verify calibration with the LCS (section 8.4) after the criteria for BFB are met (Reference 15) and prior to analysis of a blank or sample. After verification, analyze a blank (section 8.5) to demonstrate freedom from contamination and carry-over at the MDL. Tests for BFB, the LCS, and the blank are outside of the 12-hour shift, and the 12-hour shift includes samples and matrix spikes and matrix spike duplicates (section 8.4). The total time for analysis of BFB, the LCS, the blank, and the 12-hour shift must not exceed 14 hours.
12. Qualitative Identification12.1 Identification is accomplished by comparison of results from analysis of a sample or blank with data stored in the GC/MS data system (section 7.3.2.3). Identification of an analyte is confirmed per sections 12.1.1 through 12.1.4.
12.1.1 The signals for the quantitation and secondary m/z's stored in the data system (section 7.3.2.3) for each analyte of interest must be present and must maximize within the same two consecutive scans.
12.1.2 The retention time for the analyte should be within ± 10 seconds of the analyte in the LCS run at the beginning of the shift (section 8.4).
Note:Retention time windows other than ± 10 seconds may be appropriate depending on the performance of the gas chromatograph or observed retention time drifts due to certain types of matrix effects. Relative retention time (RRT) may be used as an alternative to absolute retention times if retention time drift is a concern. RRT is a unitless quantity (see section 20.2), although some procedures refer to “RRT units” in providing the specification for the agreement between the RRT values in the sample and the LCS or other standard. When significant retention time drifts are observed, dilutions or spiked samples may help the analyst determine the effects of the matrix on elution of the target analytes and to assist in qualitative identification.
12.1.3 Either the background corrected EICP areas, or the corrected relative intensities of the mass spectral peaks at the GC peak maximum, must agree within 50% to 200% ( 1/2 to 2 times) for the quantitation and secondary m/z's in the reference mass spectrum stored in the data system (section 7.3.2.3), or from a reference library. For example, if a peak has an intensity of 20% relative to the base peak, the analyte is identified if the intensity of the peak in the sample is in the range of 10% to 40% of the base peak.
12.1.4 If the acquired mass spectrum is contaminated, or if identification is ambiguous, an experienced spectrometrist (section 1.6) must determine the presence or absence of the compound.
12.2 Structural isomers that produce very similar mass spectra should be identified as individual isomers if they have sufficiently different gas chromatographic retention times. Sufficient gas chromatographic resolution is achieved if the height of the valley between two isomer peaks is less than 50% of the average of the two peak heights. Otherwise, structural isomers are identified as isomeric pairs. The resolution should be verified on the mid-point concentration of the initial calibration as well as the laboratory designated continuing calibration verification level if closely eluting isomers are to be reported.
13. Calculations13.1 When an analyte has been identified, quantitation of that analyte is based on the integrated abundance from the EICP of the primary characteristic m/z in Table 5 or 6. Calculate the concentration using the response factor (RF) determined in section 7.3.3 and Equation 2. If a calibration curve was used, calculate the concentration using the regression equation for the curve. If the concentration of an analyte exceeds the calibration range, dilute the sample by the minimum amount to bring the concentration into the calibration range, and re-analyze. Determine a dilution factor (DF) from the amount of the dilution. For example, if the extract is diluted by a factor of 2, DF = 2.
 Where: Cs
= Concentration of the analyte in the sample, and the other terms
are as defined in Section 7.3.3.
Where: Cs
= Concentration of the analyte in the sample, and the other terms
are as defined in Section 7.3.3.
13.2 Reporting of results
As noted in section 1.4.1, EPA has promulgated this method at 40 CFR part 136 for use in wastewater compliance monitoring under the National Pollutant Discharge Elimination System (NPDES). The data reporting practices described here are focused on such monitoring needs and may not be relevant to other uses of this method.
13.2.1 Report results for wastewater samples in µg/L without correction for recovery. (Other units may be used if required by a permit.) Report all QC data with the sample results.
13.2.2 Reporting level. Unless otherwise specified in by a regulatory authority or in a discharge permit, results for analytes that meet the identification criteria are reported down to the concentration of the ML established by the laboratory through calibration of the instrument (see section 7.3.2 and the glossary for the derivation of the ML). EPA considers the terms “reporting limit,” “limit of quantitation,” “quantitation limit,” and “minimum level” to be synonymous.
13.2.2.1 Report a result for each analyte in each field sample or QC standard at or above the ML to 3 significant figures. Report a result for each analyte found in each field sample or QC standard below the ML as “<ML,” where ML is the concentration of the analyte at the ML, or as required by the regulatory/control authority or permit. Report a result for each analyte in a blank at or above the MDL to 2 significant figures. Report a result for each analyte found in a blank below the MDL as “<MDL,” where MDL is the concentration of the analyte at the MDL, or as required by the regulatory/control authority or permit.
13.2.2.2 In addition to reporting results for samples and blanks separately, the concentration of each analyte in a blank associated with the sample may be subtracted from the result for that sample, but only if requested or required by a regulatory authority or in a permit. In this case, both the sample result and the blank result must be reported together.
13.2.2.3 Report a result for an analyte found in a sample that has been diluted at the least dilute level at which the area at the quantitation m/z is within the calibration range (i.e., above the ML for the analyte) and the MS/MSD recovery and RPD are within their respective QC acceptance criteria (Table 7). This may require reporting results for some analytes from different analyses.
13.2.3 Results from tests performed with an analytical system that is not in control (i.e., that does not meet acceptance criteria for any of the QC test in this method) must be documented and reported (e.g., as a qualifier on results), unless the failure is not required to be reported as determined by the regulatory/control authority. Results associated with a QC failure cannot be used to demonstrate regulatory compliance. QC failures do not relieve a discharger or permittee of reporting timely results. If the holding time would be exceeded for a re-analysis of the sample, the regulatory/control authority should be consulted for disposition.
14. Method Performance14.1 This method was tested by 15 laboratories using reagent water, drinking water, surface water, and industrial wastewaters spiked at six concentrations over the range 5-600 µg/L (References 4 and 16). Single-operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the analyte and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 8.
14.2 As noted in section 1.1, this method was validated through an interlaboratory study conducted in the early 1980s. However, the fundamental chemistry principles used in this method remain sound and continue to apply.
15. Pollution Prevention15.1 Pollution prevention encompasses any technique that reduces or eliminates the quantity or toxicity of waste at the point of generation. Many opportunities for pollution prevention exist in laboratory operations. EPA has established a preferred hierarchy of environmental management techniques that places pollution prevention as the management option of first choice. Whenever feasible, the laboratory should use pollution prevention techniques to address waste generation. When wastes cannot be reduced at the source, the Agency recommends recycling as the next best option.
15.2 The analytes in this method are used in extremely small amounts and pose little threat to the environment when managed properly. Standards should be prepared in volumes consistent with laboratory use to minimize the disposal of excess volumes of expired standards.
15.3 For information about pollution prevention that may be applied to laboratories and research institutions, consult “Less is Better: Laboratory Chemical Management for Waste Reduction,” available from the American Chemical Society's Department of Governmental Relations and Science Policy, 1155 16th Street NW., Washington, DC 20036, 202-872-4477.
16. Waste Management16.1 The laboratory is responsible for complying with all Federal, State, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions, and to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance is also required with any sewage discharge permits and regulations. An overview of requirements can be found in Environmental Management Guide for Small Laboratories (EPA 233-B-98-001).
16.2 Samples at pH <2, or pH >12, are hazardous and must be handled and disposed of as hazardous waste, or neutralized and disposed of in accordance with all federal, state, and local regulations. It is the laboratory's responsibility to comply with all federal, state, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions. The laboratory using this method has the responsibility to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance is also required with any sewage discharge permits and regulations. For further information on waste management, see “The Waste Management Manual for Laboratory Personnel,” also available from the American Chemical Society at the address in Section 15.3.
16.3 Many analytes in this method decompose above 500 °C. Low-level waste such as absorbent paper, tissues, and plastic gloves may be burned in an appropriate incinerator. Gross quantities of neat or highly concentrated solutions of toxic or hazardous chemicals should be packaged securely and disposed of through commercial or governmental channels that are capable of handling these types of wastes.
16.4 For further information on waste management, consult “Waste Management Manual for Laboratory Personnel and Less is Better-Laboratory Chemical Management for Waste Reduction,” available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street NW., Washington, DC 20036, 202-872-4477.
17. References1. Bellar, T.A. and Lichtenberg, J.J. “Determining Volatile Organics at Microgram-per-Litre Levels by Gas Chromatography,” Journal American Water Works Association, 66: 739 (1974).
2. “Sampling and Analysis Procedures for Screening of Industrial Effluents for Priority Pollutants,” U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1977, Revised April 1977.
3. Bellar, T.A. and Lichtenberg, J.J. “Semi-Automated Headspace Analysis of Drinking Waters and Industrial Waters for Purgeable Volatile Organic Compounds,” Measurement of Organic Pollutants in Water and Wastewater, C.E. Van Hall, editor, American Society for Testing and Materials, Philadelphia, PA. Special Technical Publication 686, 1978.
4. “EPA Method Study 29 EPA Method 624-Purgeables,” EPA 600/4-84-054, National Technical Information Service, PB84-209915, Springfield, Virginia 22161, June 1984.
5. 40 CFR part 136, appendix B.
6. “Method Detection Limit for Methods 624 and 625,” Olynyk, P., Budde, W.L., and Eichelberger, J.W. Unpublished report, May 14, 1980.
7. “Carcinogens-Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
8. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
9. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 7th Edition, 2003.
10. 40 CFR 136.6(b)(5)(x).
11. 40 CFR 136.6(b)(2)(i).
12. Protocol for EPA Approval of New Methods for Organic and Inorganic Analytes in Wastewater and Drinking Water (EPA-821-B-98-003) March 1999.
13. Provost, L.P. and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983).
14. 40 CFR 136.3(a), Table IB, Chlorine - Total residual.
15. Budde, W.L. and Eichelberger, J.W. “Performance Tests for the Evaluation of Computerized Gas Chromatography/Mass Spectrometry Equipment and Laboratories,” EPA-600/4-80-025, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, April 1980.
16. “Method Performance Data for Method 624,” Memorandum from R. Slater and T. Pressley, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, January 17, 1984.
18. TablesTable 1 - Purgeables 1
| Analyte | CAS Registry No. | MDL (µg/L) 2 | ML (µg/L) 3 |
|---|---|---|---|
| Acrolein | 107-02-8 | ||
| Acrylonitrile | 107-13-1 | ||
| Benzene | 71-43-2 | 4.4 | 13.2 |
| Bromodichloromethane | 75-27-4 | 2.2 | 6.6 |
| Bromoform | 75-25-2 | 4.7 | 14.1 |
| Bromomethane | 74-83-9 | ||
| Carbon tetrachloride | 56-23-5 | 2.8 | 8.4 |
| Chlorobenzene | 108-90-7 | 6.0 | 18.0 |
| Chloroethane | 75-00-3 | ||
| 2-Chloroethylvinyl ether | 110-75-8 | ||
| Chloroform | 67-66-3 | 1.6 | 4.8 |
| Chloromethane | 74-87-3 | ||
| Dibromochloromethane | 124-48-1 | 3.1 | 9.3 |
| 1,2-Dichlorobenzene | 95-50-1 | ||
| 1,3-Dichlorobenzene | 541-73-1 | ||
| 1,4-Dichlorobenzene | 106-46-7 | ||
| 1,1-Dichloroethane | 75-34-3 | 4.7 | 14.1 |
| 1,2-Dichloroethane | 107-06-2 | 2.8 | 8.4 |
| 1,1-Dichloroethene | 75-35-4 | 2.8 | 8.4 |
| trans-1,2-Dichloroethene | 156-60-5 | 1.6 | 4.8 |
| 1,2-Dichloropropane | 78-87-5 | 6.0 | 18.0 |
| cis-1,3-Dichloropropene | 10061-01-5 | 5.0 | 15.0 |
| trans-1,3-Dichloropropene | 10061-02-6 | ||
| Ethyl benzene | 100-41-4 | 7.2 | 21.6 |
| Methylene chloride | 75-09-2 | 2.8 | 8.4 |
| 1,1,2,2-Tetrachloroethane | 79-34-5 | 6.9 | 20.7 |
| Tetrachloroethene | 127-18-4 | 4.1 | 12.3 |
| Toluene | 108-88-3 | 6.0 | 18.0 |
| 1,1,1-Trichloroethane | 71-55-6 | 3.8 | 11.4 |
| 1,1,2-Trichloroethane | 79-00-5 | 5.0 | 15.0 |
| Trichloroethene | 79-01-6 | 1.9 | 5.7 |
| Vinyl chloride | 75-01-4 |
1 All the analytes in this table are Priority Pollutants (40 CFR part 423, appendix A).
2 MDL values from the 1984 promulgated version of Method 624.
3 ML = Minimum Level - see Glossary for definition and derivation.
Table 2 - Additional Purgeables
| Analyte | CAS Registry |
|---|---|
| Acetone 1 | 67-64-1 |
| Acetonitrile 2 | 75-05-8 |
| Acrolein | 107-02-8 |
| Acrylonitrile | 107-13-1 |
| Allyl alcohol 1 | 107-18-6 |
| Allyl chloride | 107-05-1 |
| t-Amyl ethyl ether (TAEE) | 919-94-8 |
| t-Amyl methyl ether (TAME) | 994-058 |
| Benzyl chloride | 100-44-7 |
| Bromoacetone 2 | 598-31-2 |
| Bromobenzene | 108-86-1 |
| Bromochloromethane | 74-97-5 |
| 1,3-Butadiene | 106-99-0 |
| n-Butanol 1 | 71-36-3 |
| 2-Butanone (MEK) 1 2 | 78-93-3 |
| t-Butyl alcohol (TBA) | 75-65-0 |
| n-Butylbenzene | 104-51-8 |
| sec-Butylbenzene | 135-98-8 |
| t-Butylbenzene | 98-06-6 |
| t-Butyl ethyl ether (ETBE) | 637-92-3 |
| Carbon disulfide | 75-15-0 |
| Chloral hydrate 2 | 302-17-0 |
| Chloroacetonitrile 1 | 107-14-2 |
| 1-Chlorobutane | 109-69-3 |
| Chlorodifluoromethane | 75-45-6 |
| 2-Chloroethanol 2 | 107-07-3 |
| bis (2-Chloroethyl) sulfide 2 | 505-60-2 |
| 1-Chlorohexanone | 20261-68-1 |
| Chloroprene (2-chloro-1,3-butadiene) | 126-99-8 |
| 3-Chloropropene | 107-05-1 |
| 3-Chloropropionitrile | 542-76-7 |
| 2-Chlorotoluene | 95-49-8 |
| 4-Chlorotoluene | 106-43-4 |
| Crotonaldehyde 1 2 | 123-73-9 |
| Cyclohexanone | 108-94-1 |
| 1,2-Dibromo-3-chloropropane | 96-12-8 |
| 1,2-Dibromoethane | 106-93-4 |
| Dibromomethane | 74-95-3 |
| cis-1,4-Dichloro-2-butene | 1476-11-5 |
| trans-1,4-Dichloro-2-butene | 110-57-6 |
| cis-1,2-Dichloroethene | 156-59-2 |
| Dichlorodifluoromethane | 75-71-8 |
| 1,3-Dichloropropane | 142-28-9 |
| 2,2-Dichloropropane | 590-20-7 |
| 1,3-Dichloro-2-propanol 2 | 96-23-1 |
| 1,1-Dichloropropene | 563-58-6 |
| cis-1,3-Dichloropropene | 10061-01-5 |
| 1:2,3:4-Diepoxybutane | 1464-53-5 |
| Diethyl ether | 60-29-7 |
| Diisopropyl ether (DIPE) | 108-20-3 |
| 1,4-Dioxane 2 | 123-91-1 |
| Epichlorohydrin 2 | 106-89-8 |
| Ethanol 2 | 64-17-5 |
| Ethyl acetate 2 | 141-78-6 |
| Ethyl methacrylate | 97-63-2 |
| Ethylene oxide 2 | 75-21-8 |
| Hexachlorobutadiene | 87-63-3 |
| Hexachloroethane | 67-72-1 |
| 2-Hexanone 2 | 591-78-6 |
| Iodomethane | 74-88-4 |
| Isobutyl alcohol 1 | 78-83-1 |
| Isopropylbenzene | 98-82-8 |
| p-Isopropyltoluene | 99-87-6 |
| Methacrylonitrile 2 | 126-98-7 |
| Methanol 2 | 67-56-1 |
| Malonitrile 2 | 109-77-3 |
| Methyl acetate | 79-20-9 |
| Methyl acrylate | 96-33-3 |
| Methyl cyclohexane | 108-87-2 |
| Methyl iodide | 74-88-4 |
| Methyl methacrylate | 78-83-1 |
| 4-Methyl-2-pentanone (MIBK) 2 | 108-10-1 |
| Methyl-t-butyl ether (MTBE) | 1634-04-4 |
| Naphthalene | 91-20-3 |
| Nitrobenzene | 98-95-3 |
| N-Nitroso-di-n-butylamine 2 | 924-16-3 |
| 2-Nitropropane | 79-46-9 |
| Paraldehyde 2 | 123-63-7 |
| Pentachloroethane 2 | 76-01-7 |
| Pentafluorobenzene | 363-72-4 |
| 2-Pentanone 2 | 107-19-7 |
| 2-Picoline 2 | 109-06-8 |
| 1-Propanol 1 | 71-23-8 |
| 2-Propanol 1 | 67-63-0 |
| Propargyl alcohol 2 | 107-19-7 |
| beta-Propiolactone 2 | 57-58-8 |
| Propionitrile (ethyl cyanide) 1 | 107-12-0 |
| n-Propylamine | 107-10-8 |
| n-Propylbenzene | 103-65-1 |
| Pyridine 2 | 110-86-1 |
| Styrene | 100-42-5 |
| 1,1,1,2-Tetrachloroethane | 630-20-6 |
| Tetrahydrofuran | 109-99-9 |
| o-Toluidine 2 | 95-53-4 |
| 1,2,3-Trichlorobenzene | 87-61-6 |
| Trichlorofluoromethane | 75-69-4 |
| 1,2,3-Trichloropropane | 96-18-4 |
| 1,2,3-Trimethylbenzene | 526-73-8 |
| 1,2,4-Trimethylbenzene | 95-63-6 |
| 1,3,5-Trimethylbenzene | 108-67-8 |
| Vinyl acetate | 108-05-4 |
| m-Xylene 3 | 108-38-3 |
| o-Xylene 3 | 95-47-6 |
| p-Xylene 3 | 106-42-3 |
| m+o-Xylene 3 | 179601-22-0 |
| m+p-Xylene 3 | 179601-23-1 |
| o+p-Xylene 3 | 136777-61-2 |
1 Determined at a purge temperature of 80 °C.
2 May be detectable at a purge temperature of 80 °C.
3 Determined in combination separated by GC column. Most GC columns will resolve o-xylene from m+p-xylene. Report using the CAS number for the individual xylene or the combination, as determined.
Table 3 - Example Retention Times
| Analyte | Retention time (min) |
|---|---|
| Chloromethane | 3.68 |
| Vinyl chloride | 3.92 |
| Bromomethane | 4.50 |
| Chloroethane | 4.65 |
| Trichlorofluoromethane | 5.25 |
| Diethyl ether | 5.88 |
| Acrolein | 6.12 |
| 1,1-Dichloroethene | 6.30 |
| Acetone | 6.40 |
| Iodomethane | 6.58 |
| Carbon disulfide | 6.72 |
| 3-Chloropropene | 6.98 |
| Methylene chloride | 7.22 |
| Acrylonitrile | 7.63 |
| trans-1,2-Dichloroethene | 7.73 |
| 1,1-Dichloroethane | 8.45 |
| Vinyl acetate | 8.55 |
| Allyl alcohol | 8.58 |
| 2-Chloro-1,3-butadiene | 8.65 |
| Methyl ethyl ketone | 9.50 |
| cis-1,2-Dichloroethene | 9.50 |
| Ethyl cyanide | 9.57 |
| Methacrylonitrile | 9.83 |
| Chloroform | 10.05 |
| 1,1,1-Trichloroethane | 10.37 |
| Carbon tetrachloride | 10.70 |
| Isobutanol | 10.77 |
| Benzene | 10.98 |
| 1,2-Dichloroethane | 11.00 |
| Crotonaldehyde | 11.45 |
| Trichloroethene | 12.08 |
| 1,2-Dichloropropane | 12.37 |
| Methyl methacrylate | 12.55 |
| p-Dioxane | 12.63 |
| Dibromomethane | 12.65 |
| Bromodichloromethane | 12.95 |
| Chloroacetonitrile | 13.27 |
| 2-Chloroethylvinyl ether | 13.45 |
| cis-1,3-Dichloropropene | 13.65 |
| 4-Methyl-2-pentanone | 13.83 |
| Toluene | 14.18 |
| trans-1,3-Dichloropropene | 14.57 |
| Ethyl methacrylate | 14.70 |
| 1,1,2-Trichloroethane | 14.93 |
| 1,3-Dichloropropane | 15.18 |
| Tetrachloroethene | 15.22 |
| 2-Hexanone | 15.30 |
| Dibromochloromethane | 15.68 |
| 1,2-Dibromoethane | 15.90 |
| Chlorobenzene | 16.78 |
| Ethylbenzene | 16.82 |
| 1,1,1,2-Tetrachloroethane | 16.87 |
| m+p-Xylene | 17.08 |
| o-Xylene | 17.82 |
| Bromoform | 18.27 |
| Bromofluorobenzene | 18.80 |
| 1,1,2,2-Tetrachloroethane | 18.98 |
| 1,2,3-Trichloropropane | 19.08 |
| trans-1,4-Dichloro-2-butene | 19.12 |
Column: 75 m x 0.53 mm ID x 3.0 µm wide-bore DB-624
Conditions: 40 °C for 4 min, 9 °C/min to 200 °C, 20 °C/min (or higher) to 250 °C, hold for 20 min at 250 °C to remove water.
Carrier gas flow rate: 6-7 mL/min at 40 °C.
Inlet split ratio: 3:1.
Interface split ratio: 7:2.
Table 4 - BFB Key m/z Abundance Criteria 1
| m/z | Abundance criteria |
|---|---|
| 50 | 15-40% of m/z 95. |
| 75 | 30-60% of m/z 95. |
| 95 | Base Peak, 100% Relative Abundance. |
| 96 | 5-9% of m/z 95. |
| 173 | <2% of m/z 174. |
| 174 | >50% of m/z 95. |
| 175 | 5-9% of m/z 174. |
| 176 | >95% but <101% of m/z 174. |
| 177 | 5-9% of m/z 176. |
1 Abundance criteria are for a quadrupole mass spectrometer. Alternative tuning criteria from other published EPA reference methods may be used, provided method performance is not adversely affected. Alternative tuning criteria specified by an instrument manufacturer may also be used for another type of mass spectrometer, or for an alternative carrier gas, provided method performance is not adversely affected.
Table 5 - Suggested Surrogate and Internal Standards
| Analyte | Retention time (min) 1 | Primary m/z | Secondary m/z's |
|---|---|---|---|
| Benzene-d6 | 10.95 | 84 | |
| 4-Bromofluorobenzene | 18.80 | 95 | 174, 176 |
| Bromochloromethane | 9.88 | 128 | 49, 130, 51 |
| 2-Bromo-1-chloropropane | 14.80 | 77 | 79, 156 |
| 2-Butanone-d5 | 9.33 | 77 | |
| Chloroethane-d5 | 4.63 | 71 | |
| Chloroform- 13C | 10.00 | 86 | |
| 1,2-Dichlorobenzene-d4 | 152 | ||
| 1,4-Dichlorobutane | 18.57 | 55 | 90, 92 |
| 1,2-Dichloroethane-d4 | 10.88 | 102 | |
| 1,1-Dichloroethene-d2 | 6.30 | 65 | |
| 1,2-Dichloropropane-d6 | 12.27 | 67 | |
| trans-1,3-Dichloropropene-d4 | 14.50 | 79 | |
| 1,4-Difluorobenzene | 114 | 63, 88 | |
| Ethylbenzene-d10 | 16.77 | 98 | |
| Fluorobenzene | 96 | 70 | |
| 2-Hexanone-d5 | 15.30 | 63 | |
| Pentafluorobenzene | 168 | ||
| 1,1,2,2-Tetrachloroethane-d2 | 18.93 | 84 | |
| Toluene-d8 | 14.13 | 100 | |
| Vinyl chloride-d3 | 3.87 | 65 |
1 For chromatographic conditions, see the footnote to Table 3.
Table 6 - Characteristic m/z's for Purgeable Organics
| Analyte | Primary m/z | Secondary m/z's |
|---|---|---|
| Acrolein | 56 | 55 and 58. |
| Acrylonitrile | 53 | 52 and 51. |
| Chloromethane | 50 | 52. |
| Bromomethane | 94 | 96. |
| Vinyl chloride | 62 | 64. |
| Chloroethane | 64 | 66. |
| Methylene chloride | 84 | 49, 51, and 86. |
| Trichlorofluoromethane | 101 | 103. |
| 1,1-Dichloroethene | 96 | 61 and 98. |
| 1,1-Dichloroethane | 63 | 65, 83, 85, 98, and 100. |
| trans-1,2-Dichloroethene | 96 | 61 and 98. |
| Chloroform | 83 | 85. |
| 1,2-Dichloroethane | 98 | 62, 64, and 100. |
| 1,1,1-Trichloroethane | 97 | 99, 117, and 119. |
| Carbon tetrachloride | 117 | 119 and 121. |
| Bromodichloromethane | 83 | 127, 85, and 129. |
| 1,2-Dichloropropane | 63 | 112, 65, and 114. |
| trans-1,3-Dichloropropene | 75 | 77. |
| Trichloroethene | 130 | 95, 97, and 132. |
| Benzene | 78 | |
| Dibromochloromethane | 127 | 129, 208, and 206. |
| 1,1,2-Trichloroethane | 97 | 83, 85, 99, 132, and 134. |
| cis-1,3-Dichloropropene | 75 | 77. |
| 2-Chloroethylvinyl ether | 106 | 63 and 65. |
| Bromoform | 173 | 171, 175, 250, 252, 254, and 256. |
| 1,1,2,2-Tetrachloroethane | 168 | 83, 85, 131, 133, and 166. |
| Tetrachloroethene | 164 | 129, 131, and 166. |
| Toluene | 92 | 91. |
| Chlorobenzene | 112 | 114. |
| Ethyl benzene | 106 | 91. |
| 1,3-Dichlorobenzene | 146 | 148 and 111. |
| 1,2-Dichlorobenzene | 146 | 148 and 111. |
| 1,4-Dichlorobenzene | 146 | 148 and 111. |
Table 7 - LCS (Q), DOC (s and X), and MS/MSD (P and RPD) Acceptance Criteria 1
| Analyte | Range for Q (%) |
Limit for s (%) |
Range for X (%) |
Range for P1, P2 (%) |
Limit for RPD |
|---|---|---|---|---|---|
| Acrolein | 60-140 | 30 | 50-150 | 40-160 | 60 |
| Acrylonitrile | 60-140 | 30 | 50-150 | 40-160 | 60 |
| Benzene | 65-135 | 33 | 75-125 | 37-151 | 61 |
| Benzene-d6 | |||||
| Bromodichloromethane | 65-135 | 34 | 50-140 | 35-155 | 56 |
| Bromoform | 70-130 | 25 | 57-156 | 45-169 | 42 |
| Bromomethane | 15-185 | 90 | D-206 | D-242 | 61 |
| 2-Butanone-d5 | |||||
| Carbon tetrachloride | 70-130 | 26 | 65-125 | 70-140 | 41 |
| Chlorobenzene | 65-135 | 29 | 82-137 | 37-160 | 53 |
| Chloroethane | 40-160 | 47 | 42-202 | 14-230 | 78 |
| Chloroethane-d5 | |||||
| 2-Chloroethylvinyl ether | D-225 | 130 | D-252 | D-305 | 71 |
| Chloroform | 70-135 | 32 | 68-121 | 51-138 | 54 |
| Chloroform- 13C | |||||
| Chloromethane | D-205 | 472 | D-230 | D-273 | 60 |
| Dibromochloromethane | 70-135 | 30 | 69-133 | 53-149 | 50 |
| 1,2-Dichlorobenzene | 65-135 | 31 | 59-174 | 18-190 | 57 |
| 1,2-Dichlorobenzene-d4 | |||||
| 1,3-Dichlorobenzene | 70-130 | 24 | 75-144 | 59-156 | 43 |
| 1,4-Dichlorobenzene | 65-135 | 31 | 59-174 | 18-190 | 57 |
| 1,1-Dichloroethane | 70-130 | 24 | 71-143 | 59-155 | 40 |
| 1,2-Dichloroethane | 70-130 | 29 | 72-137 | 49-155 | 49 |
| 1,2-Dichloroethane-d4 | |||||
| 1,1-Dichloroethene | 50-150 | 40 | 19-212 | D-234 | 32 |
| 1,1-Dichloroethene-d2 | |||||
| trans-1,2-Dichloroethene | 70-130 | 27 | 68-143 | 54-156 | 45 |
| 1,2-Dichloropropane | 35-165 | 69 | 19-181 | D-210 | 55 |
| 1,2-Dichloropropane-d6 | |||||
| cis-1,3-Dichloropropene | 25-175 | 79 | 5-195 | D-227 | 58 |
| trans-1,3-Dichloropropene | 50-150 | 52 | 38-162 | 17-183 | 86 |
| trans-1,3-Dichloropropene-d4 | |||||
| Ethyl benzene | 60-140 | 34 | 75-134 | 37-162 | 63 |
| 2-Hexanone-d5 | |||||
| Methylene chloride | 60-140 | 192 | D-205 | D-221 | 28 |
| 1,1,2,2-Tetrachloroethane | 60-140 | 36 | 68-136 | 46-157 | 61 |
| 1,1,2,2-Tetrachloroethane-d2 | |||||
| Tetrachloroethene | 70-130 | 23 | 65-133 | 64-148 | 39 |
| Toluene | 70-130 | 22 | 75-134 | 47-150 | 41 |
| Toluene-d8 | |||||
| 1,1,1-Trichloroethane | 70-130 | 21 | 69-151 | 52-162 | 36 |
| 1,1,2-Trichloroethane | 70-130 | 27 | 75-136 | 52-150 | 45 |
| Trichloroethene | 65-135 | 29 | 75-138 | 70-157 | 48 |
| Trichlorofluoromethane | 50-150 | 50 | 45-158 | 17-181 | 84 |
| Vinyl chloride | 5-195 | 100 | D-218 | D-251 | 66 |
| Vinyl chloride-d3 |
1 Criteria were calculated using an LCS concentration of 20 µg/L.
Q = Percent recovery in calibration verification/LCS (section 8.4).
s = Standard deviation of percent recovery for four recovery measurements (section 8.2.4).
X = Average percent recovery for four recovery measurements (section 8.2.4).
P = Percent recovery for the MS or MSD (section 8.3.3).
D = Detected; result must be greater than zero.
Notes:
1. Criteria for pollutants are based upon the method performance data in Reference 4. Where necessary, limits have been broadened to assure applicability to concentrations below those used to develop Table 7.
2. Criteria for surrogates are from EPA CLP SOM01.2D.
Table 8 - Recovery and Precision as Functions of Concentration
| Analyte | Recovery, X′ (µg/L) |
Single analyst precision,
sr′ (µg/L) |
Overall precision, S′ (µg/L) |
|---|---|---|---|
| Benzene | 0.93C+2.00 | 20.26 X−1.74 | 0.25 X−1.33 |
| Bromodichloromethane | 1.03C−1.58 | 0.15 X+0.59 | 0.20 X+1.13 |
| Bromoform | 1.18C−2.35 | 0.12 X+0.36 | 0.17 X+1.38 |
| Bromomethane a | 1.00C | 0.43 X | 0.58 X |
| Carbon tetrachloride | 1.10C−1.68 | 0.12 X+0.25 | 0.11 X+0.37 |
| Chlorobenzene | 0.98C+2.28 | 0.16 X−0.09 | 0.26 X−1.92 |
| Chloroethane | 1.18C+0.81 | 0.14 X+2.78 | 0.29 X+1.75 |
| 2-Chloroethylvinyl ether a | 1.00C | 0.62 X | 0.84 X |
| Chloroform | 0.93C+0.33 | 0.16 X+0.22 | 0.18 X+0.16 |
| Chloromethane | 1.03C+0.81 | 0.37 X+2.14 | 0.58 X+0.43 |
| Dibromochloromethane | 1.01C−0.03 | 0.17 X−0.18 | 0.17 X+0.49 |
| 1,2-Dichlorobenzene b | 0.94C+4.47 | 0.22 X−1.45 | 0.30 X−1.20 |
| 1,3-Dichlorobenzene | 1.06C+1.68 | 0.14 X−0.48 | 0.18 X−0.82 |
| 1,4-Dichlorobenzene b | 0.94C+4.47 | 0.22 X−1.45 | 0.30 X−1.20 |
| 1,1-Dichloroethane | 1.05C+0.36 | 0.13 X−0.05 | 0.16 X+0.47 |
| 1,2-Dichloroethane | 1.02C+0.45 | 0.17 X−0.32 | 0.21 X−0.38 |
| 1,1-Dichloroethene | 1.12C+0.61 | 0.17 X+1.06 | 0.43 X−0.22 |
| trans-1,2,-Dichloroethene | 1.05C+0.03 | 0.14 X−+0.09 | 0.19 X−+0.17 |
| 1,2-Dichloropropane a | 1.00C | 0.33 X | 0.45 X |
| cis-1,3-Dichloropropene a | 1.00C | 0.38 X | 0.52 X |
| trans-1,3-Dichloropropene a | 1.00C | 0.25 X | 0.34 X |
| Ethyl benzene | 0.98C+2.48 | 0.14 X+1.00 | 0.26 X−1.72 |
| Methylene chloride | 0.87C+1.88 | 0.15 X+1.07 | 0.32 X+4.00 |
| 1,1,2,2-Tetrachloroethane | 0.93C+1.76 | 0.16 X+0.69 | 0.20 X+0.41 |
| Tetrachloroethene | 1.06C+0.60 | 0.13 X−0.18 | 0.16 X−0.45 |
| Toluene | 0.98C+2.03 | 0.15 X−0.71 | 0.22 X−1.71 |
| 1,1,1-Trichloroethane | 1.06C+0.73 | 0.12 X−0.15 | 0.21 X−0.39 |
| 1,1,2-Trichloroethane | 0.95C+1.71 | 0.14 X+0.02 | 0.18 X+0.00 |
| Trichloroethene | 1.04C+2.27 | 0.13 X+0.36 | 0.12 X+0.59 |
| Trichlorofluoromethane | 0.99C+0.39 | 0.33 X−1.48 | 0.34 X−0.39 |
| Vinyl chloride | 1.00C | 0.48 X | 0.65 X |
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
Sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X, in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X, in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
a Estimates based upon the performance in a single laboratory (References 4 and 16).
b Due to coelutions, performance statements for these isomers are based upon the sums of their concentrations.
These definitions and purposes are specific to this method, but have been conformed to common usage to the extent possible.
19.1 Units of weight and measure and their abbreviations.
19.1.1 Symbols.
°C degrees Celsius µg microgram µL microliter < less than > greater than % percent19.1.2 Abbreviations (in alphabetical order).
cm centimeter g gram h hour ID inside diameter in. inch L liter m mass mg milligram min minute mL milliliter mm millimeter ms millisecond m/z mass-to-charge ratio N normal; gram molecular weight of solute divided by hydrogen equivalent of solute, per liter of solution ng nanogram pg picogram ppb part-per-billion ppm part-per-million ppt part-per-trillion psig pounds-per-square inch gauge v/v volume per unit volume w/v weight per unit volume19.2 Definitions and acronyms (in alphabetical order).
Analyte - A compound tested for by this method. The analytes are listed in Tables 1 and 2.
Analyte of interest - An analyte of interest is an analyte required to be determined by a regulatory/control authority or in a permit, or by a client.
Analytical batch - The set of samples analyzed on a given instrument during a 12-hour period that begins with analysis of a calibration verification/LCS. See section 8.4.
Blank - An aliquot of reagent water that is treated exactly as a sample including exposure to all glassware, equipment, solvents, reagents, internal standards, and surrogates that are used with samples. The blank is used to determine if analytes or interferences are present in the laboratory environment, the reagents, or the apparatus. See section 8.5.
Calibration - The process of determining the relationship between the output or response of a measuring instrument and the value of an input standard. Historically, EPA has referred to a multi-point calibration as the “initial calibration,” to differentiate it from a single-point calibration verification.
Calibration standard - A solution prepared from stock solutions and/or a secondary standards and containing the analytes of interest, surrogates, and internal standards. The calibration standard is used to calibrate the response of the GC/MS instrument against analyte concentration.
Calibration verification standard - The laboratory control sample (LCS) used to verify calibration. See Section 8.4.
Descriptor - In SIM, the beginning and ending retention times for the RT window, the m/z's sampled in the RT window, and the dwell time at each m/z.
Extracted ion current profile (EICP) - The line described by the signal at a given m/z.
Field duplicates - Two samples collected at the same time and place under identical conditions, and treated identically throughout field and laboratory procedures. Results of analyses of field duplicates provide an estimate of the precision associated with sample collection, preservation, and storage, as well as with laboratory procedures.
Field blank - An aliquot of reagent water or other reference matrix that is placed in a sample container in the field, and treated as a sample in all respects, including exposure to sampling site conditions, storage, preservation, and all analytical procedures. The purpose of the field blank is to determine if the field or sample transporting procedures and environments have contaminated the sample.
GC - Gas chromatograph or gas chromatography.
Internal standard - A compound added to a sample in a known amount and used as a reference for quantitation of the analytes of interest and surrogates. Internal standards are listed in Table 5. Also see Internal standard quantitation.
Internal standard quantitation - A means of determining the concentration of an analyte of interest (Tables 1 and 2) by reference to a compound added to a sample and not expected to be found in the sample.
DOC - Initial demonstration of capability (DOC; section 8.2); four aliquots of reagent water spiked with the analytes of interest and analyzed to establish the ability of the laboratory to generate acceptable precision and recovery. A DOC is performed prior to the first time this method is used and any time the method or instrumentation is modified.
Laboratory control sample (LCS; laboratory fortified blank (LFB); on-going precision and recovery sample; OPR) - An aliquot of reagent water spiked with known quantities of the analytes of interest and surrogates. The LCS is analyzed exactly like a sample. Its purpose is to assure that the results produced by the laboratory remain within the limits specified in this method for precision and recovery. In this method, the LCS is synonymous with a calibration verification sample (See sections 7.4 and 8.4).
Laboratory fortified sample matrix - See Matrix spike.
Laboratory reagent blank - See Blank.
Matrix spike (MS) and matrix spike duplicate (MSD) (laboratory fortified sample matrix and duplicate) - Two aliquots of an environmental sample to which known quantities of the analytes of interest and surrogates are added in the laboratory. The MS/MSD are prepared and analyzed exactly like a field sample. Their purpose is to quantify any additional bias and imprecision caused by the sample matrix. The background concentrations of the analytes in the sample matrix must be determined in a separate aliquot and the measured values in the MS/MSD corrected for background concentrations.
May - This action, activity, or procedural step is neither required nor prohibited.
May not - This action, activity, or procedural step is prohibited.
Method blank (laboratory reagent blank) - See Blank.
Method detection limit (MDL) - A detection limit determined by the procedure at 40 CFR part 136, appendix B. The MDLs determined by EPA in the original version of the method are listed in Table 1. As noted in Sec. 1.4, use the MDLs in Table 1 in conjunction with current MDL data from the laboratory actually analyzing samples to assess the sensitivity of this procedure relative to project objectives and regulatory requirements (where applicable).
Minimum level (ML) - The term “minimum level” refers to either the sample concentration equivalent to the lowest calibration point in a method or a multiple of the method detection limit (MDL), whichever is higher. Minimum levels may be obtained in several ways: They may be published in a method; they may be based on the lowest acceptable calibration point used by a laboratory; or they may be calculated by multiplying the MDL in a method, or the MDL determined by a laboratory, by a factor of 3. For the purposes of NPDES compliance monitoring, EPA considers the following terms to be synonymous: “quantitation limit,” “reporting limit,” and “minimum level.”
MS - Mass spectrometer or mass spectrometry.
Must - This action, activity, or procedural step is required.
m/z - The ratio of the mass of an ion (m) detected in the mass spectrometer to the charge (z) of that ion.
Quality control sample (QCS) - A sample containing analytes of interest at known concentrations. The QCS is obtained from a source external to the laboratory or is prepared from standards obtained from a different source than the calibration standards.
The purpose is to check laboratory performance using test materials that have been prepared independent of the normal preparation process.
Reagent water - Water demonstrated to be free from the analytes of interest and potentially interfering substances at the MDLs for the analytes in this method.
Regulatory compliance limit (or regulatory concentration limit) - A limit on the concentration or amount of a pollutant or contaminant specified in a nationwide standard, in a permit, or otherwise established by a regulatory/control authority.
Relative retention time (RRT) - The ratio of the retention time of an analyte to the retention time of its associated internal standard. RRT compensates for small changes in the GC temperature program that can affect the absolute retention times of the analyte and internal standard. RRT is a unitless quantity.
Relative standard deviation (RSD) - The standard deviation times 100 divided by the mean. Also termed “coefficient of variation.”
RF - Response factor. See section 7.3.3.
RSD - See relative standard deviation.
Safety Data Sheet (SDS) - Written information on a chemical's toxicity, health hazards, physical properties, fire, and reactivity, including storage, spill, and handling precautions that meet the requirements of OSHA, 29 CFR 1910.1200(g) and appendix D to § 1910.1200. United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS), third revised edition, United Nations, 2009.
Selected Ion Monitoring (SIM) - An MS technique in which a few m/z's are monitored. When used with gas chromatography, the m/z's monitored are usually changed periodically throughout the chromatographic run to correlate with the characteristic m/z's for the analytes, surrogates, and internal standards as they elute from the chromatographic column. The technique is often used to increase sensitivity and minimize interferences.
Signal-to-noise ratio (S/N) - The height of the signal as measured from the mean (average) of the noise to the peak maximum divided by the width of the noise.
SIM - See Selection Ion Monitoring.
Should - This action, activity, or procedural step is suggested but not required.
Stock solution - A solution containing an analyte that is prepared using a reference material traceable to EPA, the National Institute of Science and Technology (NIST), or a source that will attest to the purity and authenticity of the reference material.
Surrogate - A compound unlikely to be found in a sample, and which is spiked into sample in a known amount before purge-and-trap. The surrogate is quantitated with the same procedures used to quantitate the analytes of interest. The purpose of the surrogate is to monitor method performance with each sample.
VOA - Volatile organic analysis: e.g., the analysis performed by this method.
Method 625.1 - Base/Neutrals and Acids by GC/MS 1. Scope and Application1.1 This method is for determination of semivolatile organic pollutants in industrial discharges and other environmental samples by gas chromatography combined with mass spectrometry (GC/MS), as provided under 40 CFR 136.1. This revision is based on a previous protocol (Reference 1), on the basic revision promulgated October 26, 1984, and on an interlaboratory method validation study (Reference 2). Although this method was validated through an interlaboratory study conducted in the early 1980s, the fundamental chemistry principles used in this method remain sound and continue to apply.
1.2 The analytes that may be qualitatively and quantitatively determined using this method and their CAS Registry numbers are listed in Tables 1 and 2. The method may be extended to determine the analytes listed in Table 3; however, extraction or gas chromatography of some of these analytes may make quantitative determination difficult. For example, benzidine is subject to oxidative losses during extraction and/or solvent concentration. Under the alkaline conditions of the extraction, alpha-BHC, gamma-BHC, endosulfan I and II, and endrin are subject to decomposition. Hexachlorocyclopentadiene is subject to thermal decomposition in the inlet of the gas chromatograph, chemical reaction in acetone solution, and photochemical decomposition. N-nitrosodiphenylamine and other nitrosoamines may decompose in the gas chromatographic inlet. The sample may be extracted at neutral pH if necessary to overcome these or other decomposition problems that could occur at alkaline or acidic pH. EPA also has provided other methods (e.g., Method 607 - Nitrosamines) that may be used for determination of some of these analytes. EPA encourages use of Method 625.1 to determine additional compounds amenable to extraction and GC/MS.
1.3 The large number of analytes in Tables 1-3 of this method makes testing difficult if all analytes are determined simultaneously. Therefore, it is necessary to determine and perform quality control (QC) tests for the “analytes of interest” only. Analytes of interest are those required to be determined by a regulatory/control authority or in a permit, or by a client. If a list of analytes is not specified, the analytes in Tables 1 and 2 must be determined, at a minimum, and QC testing must be performed for these analytes. The analytes in Tables 1 and 2, and some of the analytes in Table 3 have been identified as Toxic Pollutants (40 CFR 401.15), expanded to a list of Priority Pollutants (40 CFR part 423, appendix A).
1.4 In this revision to Method 625, the pesticides and polychlorinated biphenyls (PCBs) have been moved from Table 1 to Table 3 (Additional Analytes) to distinguish these analytes from the analytes required in quality control tests (Tables 1 and 2). QC acceptance criteria for pesticides and PCBs have been retained in Table 6 and may continue to be applied if desired, or if requested or required by a regulatory/control authority or in a permit. Method 608.3 should be used for determination of pesticides and PCBs. However, if pesticides and/or PCBs are to be determined, an additional sample must be collected and extracted using the pH adjustment and extraction procedures specified in Method 608.3. Method 1668C may be useful for determination of PCBs as individual chlorinated biphenyl congeners, and Method 1699 may be useful for determination of pesticides. At the time of writing of this revision, Methods 1668C and 1699 had not been approved for use at 40 CFR part 136. The screening procedure for 2,3,7,8-tetrachlorodibenzo-p-dioxin (2,3,7,8-TCDD) contained in the version of Method 625 promulgated October 26, 1984 has been replaced with procedures for selected ion monitoring (SIM), and 2,3,7,8-TCDD may be determined using the SIM procedures. However, EPA Method 613 or 1613B should be used for analyte-specific determination of 2,3,7,8-TCDD because of the focus of these methods on this compound. Methods 613 and 1613B are approved for use at 40 CFR part 136.
1.5 Method detection limits (MDLs; Reference 3) for the analytes in Tables 1, 2, and 3 are listed in those tables. These MDLs were determined in reagent water (Reference 4). Advances in analytical technology, particularly the use of capillary (open-tubular) columns, allowed laboratories to routinely achieve MDLs for the analytes in this method that are 2-10 times lower than those in the version promulgated in 1984. The MDL for an analyte in a specific wastewater may differ from those listed, depending upon the nature of interferences in the sample matrix.
1.5.1 EPA has promulgated this method at 40 CFR part 136 for use in wastewater compliance monitoring under the National Pollutant Discharge Elimination System (NPDES). The data reporting practices described in section 15.2 are focused on such monitoring needs and may not be relevant to other uses of the method.
1.5.2 This method includes “reporting limits” based on EPA's “minimum level” (ML) concept (see the glossary in section 22). Tables 1, 2, and 3 contain MDL values and ML values for many of the analytes.
1.6 This method is performance-based. It may be modified to improve performance (e.g., to overcome interferences or improve the accuracy of results) provided all performance requirements are met.
1.6.1 Examples of allowed method modifications are described at 40 CFR 136.6. Other examples of allowed modifications specific to this method, including solid-phase extraction (SPE) are described in section 8.1.2.
1.6.2 Any modification beyond those expressly permitted at 40 CFR 136.6 or in section 8.1.2 of this method shall be considered a major modification subject to application and approval of an alternate test procedure under 40 CFR 136.4 and 136.5.
1.6.3 For regulatory compliance, any modification must be demonstrated to produce results equivalent or superior to results produced by this method when applied to relevant wastewaters (section 8.3).
1.7 This method is restricted to use by or under the supervision of analysts experienced in the use of a gas chromatograph/mass spectrometer and in the interpretation of mass spectra. Each laboratory that uses this method must demonstrate the ability to generate acceptable results using the procedure in Section 8.2.
1.8 Terms and units of measure used in this method are given in the glossary at the end of the method.
2. Summary of Method2.1 A measured volume of sample, sufficient to meet an MDL or reporting limit, is serially extracted with methylene chloride at pH 11-13 and again at a pH less than 2 using a separatory funnel or continuous liquid/liquid extractor.
2.2 The extract is concentrated to a volume necessary to meet the required compliance or detection limit, and analyzed by GC/MS. Qualitative identification of an analyte in the extract is performed using the retention time and the relative abundance of two or more characteristic masses (m/z's). Quantitative analysis is performed using the internal standard technique with a single characteristic m/z.
3. Contamination and Interferences3.1 Solvents, reagents, glassware, and other sample processing labware may yield artifacts, elevated baselines, or matrix interferences causing misinterpretation of chromatograms and mass spectra. All materials used in the analysis must be demonstrated to be free from contamination and interferences by analyzing blanks initially and with each extraction batch (samples started through the extraction process in a given 24-hour period, to a maximum of 20 samples - see Glossary for detailed definition), as described in Section 8.5. Specific selection of reagents and purification of solvents by distillation in all-glass systems may be required. Where possible, labware is cleaned by extraction or solvent rinse, or baking in a kiln or oven.
3.2 Glassware must be scrupulously cleaned (Reference 5). Clean all glassware as soon as possible after use by rinsing with the last solvent used in it. Solvent rinsing should be followed by detergent washing with hot water, and rinses with tap water and reagent water. The glassware should then be drained dry, and heated at 400 °C for 15-30 minutes. Some thermally stable materials, such as PCBs, may require higher temperatures and longer baking times for removal. Solvent rinses with pesticide quality acetone, hexane, or other solvents may be substituted for heating. Do not heat volumetric labware above 90 °C. After drying and cooling, store inverted or capped with solvent-rinsed or baked aluminum foil in a clean environment to prevent accumulation of dust or other contaminants.
3.3 Matrix interferences may be caused by contaminants co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the nature and diversity of the industrial complex or municipality being sampled. Interferences extracted from samples high in total organic carbon (TOC) may result in elevated baselines, or by enhancing or suppressing a signal at or near the retention time of an analyte of interest. Analyses of the matrix spike and duplicate (section 8.3) may be useful in identifying matrix interferences, and gel permeation chromatography (GPC; Section 11.1) and sulfur removal (section 11.2) may aid in eliminating these interferences. EPA has provided guidance that may aid in overcoming matrix interferences (Reference 6).
3.4 In samples that contain an inordinate number of interferences, the use of chemical ionization (CI) or triple quadrupole (MRM) mass spectrometry may make identification easier. Tables 4 and 5 give characteristic CI and MRM m/z's for many of the analytes covered by this method. The use of CI or MRM mass spectrometry may be utilized to support electron ionization (EI) mass spectrometry or as a primary method for identification and quantification. While the use of these enhanced techniques is encouraged, it is not required.
4. Safety4.1 Hazards associated with each reagent used in this method have not been precisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of safety data sheets (SDSs, OSHA, 29 CFR 1910.1200(g)) should also be made available to all personnel involved in sample handling and chemical analysis. Additional references to laboratory safety are available and have been identified (References 7-9) for the information of the analyst.
4.2 The following analytes covered by this method have been tentatively classified as known or suspected human or mammalian carcinogens: Benzo(a)anthracene, benzidine, 3,3′-dichlorobenzidine, benzo(a)pyrene, alpha-BHC, beta-BHC, delta-BHC, gamma-BHC, Dibenz(a,h)-anthracene, N-nitrosodimethylamine, 4,4′-DDT, and PCBs. Other compounds in Table 3 may also be toxic. Primary standards of toxic compounds should be prepared in a chemical fume hood, and a NIOSH/MESA approved toxic gas respirator should be worn when handling high concentrations of these compounds.
4.3 This method allows the use of hydrogen as a carrier gas in place of helium (section 5.6.1.2). The laboratory should take the necessary precautions in dealing with hydrogen, and should limit hydrogen flow at the source to prevent buildup of an explosive mixture of hydrogen in air.
5. Apparatus and Materials Note:Brand names, suppliers, and part numbers are for illustration purposes only. No endorsement is implied. Equivalent performance may be achieved using equipment and materials other than those specified here. Demonstrating that the equipment and supplies used in the laboratory achieves the required performance is the responsibility of the laboratory. Suppliers for equipment and materials in this method may be found through an on-line search. Please do not contact EPA for supplier information.
5.1 Sampling equipment, for discrete or composite sampling.
5.1.1 Grab sample bottle - amber glass bottle large enough to contain the necessary sample volume, fitted with a fluoropolymer-lined screw cap. Foil may be substituted for fluoropolymer if the sample is not corrosive. If amber bottles are not available, protect samples from light. Unless pre-cleaned, the bottle and cap liner must be washed, rinsed with acetone or methylene chloride, and dried before use to minimize contamination.
5.1.2 Automatic sampler (optional) - the sampler must incorporate a pre-cleaned glass sample container. Samples must be kept refrigerated at ≤6 °C and protected from light during compositing. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used. Before use, however, rinse the compressible tubing with methanol, followed by repeated rinsing with reagent water, to minimize the potential for sample contamination. An integrating flow meter is required to collect flow-proportioned composites.
5.2 Glassware.
5.2.1 Separatory funnel - Size appropriate to hold sample volume and extraction solvent volume, and equipped with fluoropolymer stopcock.
5.2.2 Drying column - Chromatographic column, approximately 400 mm long by 19 mm ID, with coarse frit, or equivalent, sufficient to hold 15 g of anhydrous sodium sulfate.
5.2.3 Concentrator tube, Kuderna-Danish - 10 mL, graduated (Kontes 570050-1025 or equivalent). Calibration must be checked at the volumes employed in the test. A ground glass stopper is used to prevent evaporation of extracts.
5.2.4 Evaporative flask, Kuderna-Danish - 500 mL (Kontes 57001-0500 or equivalent). Attach to concentrator tube with springs.
Note:Use of a solvent recovery system with the K-D or other solvent evaporation apparatus is strongly recommended.
5.2.5 Snyder column, Kuderna-Danish - Three-ball macro (Kontes 503000-0121 or equivalent).
5.2.6 Snyder column, Kuderna-Danish - Two-ball micro (Kontes 569001-0219 or equivalent).
5.2.7 Vials - 10-15 mL, amber glass, with Teflon-lined screw cap.
5.2.8 Continuous liquid-liquid extractor - Equipped with fluoropolymer or glass connecting joints and stopcocks requiring no lubrication. (Hershberg-Wolf Extractor, Ace Glass Company, Vineland, NJ, P/N 6848-20, or equivalent.)
5.2.9 In addition to the glassware listed above, the laboratory should be equipped with all necessary pipets, volumetric flasks, beakers, and other glassware listed in this method and necessary to perform analyses successfully.
5.3 Boiling chips - Approximately 10/40 mesh, glass, silicon carbide, or equivalent. Heat to 400 °C for 30 minutes, or solvent rinse or Soxhlet extract with methylene chloride.
5.4 Water bath - Heated, with concentric ring cover, capable of temperature control (±2 °C). The bath should be used in a hood.
5.5 Balances.
5.5.1 Analytical, capable of accurately weighing 0.1 mg.
5.5.2 Top loading, capable of accurately weighing 10 mg.
5.6 GC/MS system.
5.6.1 Gas chromatograph (GC) - An analytical system complete with a temperature programmable gas chromatograph and all required accessories, including syringes and analytical columns.
5.6.1.1 Injection port - Can be split, splitless, temperature programmable vaporization split/splitless (PTV), solvent-purge, large-volume, on-column, backflushed, or other. An autosampler is highly recommended because it injects volumes more precisely than volumes injected manually.
5.6.1.2 Carrier gas - Helium or hydrogen. Data in the tables in this method were obtained using helium carrier gas. If hydrogen is used, analytical conditions may need to be adjusted for optimum performance, and calibration and all QC tests must be performed with hydrogen carrier gas. See Section 4.3 for precautions regarding the use of hydrogen as a carrier gas.
5.6.2 GC column - See the footnotes to Tables 4 and 5. Other columns or column systems may be used provided all requirements in this method are met.
5.6.3 Mass spectrometer - Capable of repetitively scanning from 35-450 Daltons (amu) every two seconds or less, utilizing a 70 eV (nominal) electron energy in the electron impact ionization mode, and producing a mass spectrum which meets all the criteria in Table 9A or 9B when 50 ng or less of decafluorotriphenyl phosphine (DFTPP; CAS 5074-71-5; bis(pentafluorophenyl) phenyl phosphine) is injected into the GC.
5.6.4 GC/MS interface - Any GC to MS interface that meets all performance requirements in this method may be used.
5.6.5 Data system - A computer system must be interfaced to the mass spectrometer that allows the continuous acquisition and storage of mass spectra acquired throughout the chromatographic program. The computer must have software that allows searching any GC/MS data file for specific m/z's (masses) and plotting m/z abundances versus time or scan number. This type of plot is defined as an extracted ion current profile (EICP). Software must also be available that allows integrating the abundance at any EICP between specified time or scan number limits.
5.7 Automated gel permeation chromatograph (GPC).
5.7.1 GPC column - 150-700 mm long × 21-25 mm ID, packed with 70 g of SX-3 Biobeads; Bio-Rad Labs, or equivalent.
5.7.2 Pump, injection valve, UV detector, and other apparatus necessary to meet the requirements in this method.
5.8 Nitrogen evaporation device - Equipped with a water bath than can be maintained at 30-45 °C; N-Evap, Organomation Associates, or equivalent.
5.9 Muffle furnace or kiln - Capable of baking glassware or sodium sulfate in the range of 400-450 °C.
6. Reagents6.1 Reagent water - Reagent water is defined as water in which the analytes of interest and interfering compounds are not detected at the MDLs of the analytes of interest.
6.2 Sodium hydroxide solution (10 N) - Dissolve 40 g of NaOH (ACS) in reagent water and dilute to 100 mL.
6.3 Sodium thiosulfate - (ACS) granular.
6.4 Sulfuric acid (1+1) - Slowly add 50 mL of H2SO4 (ACS, sp. gr. 1.84) to 50 mL of reagent water.
6.5 Acetone, methanol, methylene chloride, 2-propanol - High purity pesticide quality, or equivalent, demonstrated to be free of the analytes of interest and interferences (Section 3). Purification of solvents by distillation in all-glass systems may be required.
6.6 Sodium sulfate - (ACS) granular, anhydrous, rinsed or Soxhlet extracted with methylene chloride (20 mL/g), baked in a shallow tray at 450 °C for one hour minimum, cooled in a desiccator, and stored in a pre-cleaned glass bottle with screw cap that prevents moisture from entering.
6.7 Stock standard solutions (1.00 µg/µL) - Stock standard solutions may be prepared from pure materials, or purchased as certified solutions. Traceability must be to the National Institute of Standards and Technology (NIST) or other national or international standard, when available. Stock solution concentrations alternate to those below may be used. Because of the toxicity of some of the compounds, primary dilutions should be prepared in a hood, and a NIOSH/MESA approved toxic gas respirator should be worn when high concentrations of neat materials are handled. The following procedure may be used to prepare standards from neat materials.
6.7.1 Prepare stock standard solutions by accurately weighing about 0.0100 g of pure material. Dissolve the material in pesticide quality methanol or other suitable solvent and dilute to volume in a 10-mL volumetric flask. Larger volumes may be used at the convenience of the laboratory. When compound purity is assayed to be 96% or greater, the weight may be used without correction to calculate the concentration of the stock standard. Commercially prepared stock standards may be used at any concentration if they are certified by the manufacturer or by an independent source.
6.7.2 Unless stated otherwise in this method, store non-aqueous standards in fluoropolymer-lined screw-cap, or heat-sealed, glass containers, in the dark at −20 to −10 °C. Store aqueous standards; e.g., the aqueous LCS (section 8.4.1), in the dark at ≤ 6 °C, but do not freeze. Standards prepared by the laboratory may be stored for up to one year, except when comparison with QC check standards indicates that a standard has degraded or become more concentrated due to evaporation, or unless the laboratory has data on file to prove stability for a longer period. Commercially prepared standards may be stored until the expiration date provided by the vendor, except when comparison with QC check standards indicates that a standard has degraded or become more concentrated due to evaporation, or unless the laboratory has data from the vendor on file to prove stability for a longer period.
6.8 Surrogate standard spiking solution.
6.8.1 Select a minimum of three surrogate compounds from Table 8 that most closely match the recovery of the analytes of interest. For example, if all analytes tested are considered acids, use surrogates that have similar chemical attributes. Other compounds may be used as surrogates so long as they do not interfere in the analysis. If only one or two analytes are determined, one or two surrogates may be used.
6.8.2 Prepare a solution containing each selected surrogate such that the concentration in the sample would match the concentration in the mid-point calibration standard. For example, if the midpoint of the calibration is 100 µg/L, prepare the spiking solution at a concentration of 100 µg/mL in methanol. Addition of 1.00 mL of this solution to 1000 mL of sample will produce a concentration of 100 µg/L of the surrogate. Alternate volumes and concentrations appropriate to the response of the GC/MS instrument or for selective ion monitoring (SIM) may be used, if desired. Store per section 6.7.2.
6.9 Internal standard spiking solution.
6.9.1 Select three or more internal standards similar in chromatographic behavior to the analytes of interest. Internal standards are listed in Table 8. Suggested internal standards are: 1,4-dichlorobenzene-d4; naphthalene-d8; acenaphthene-d10; phenanthrene-d10; chrysene-d12; and perylene-d12. The laboratory must demonstrate that measurement of the internal standards is not affected by method or matrix interferences (see also section 7.3.4).
6.9.2 Prepare the internal standards at a concentration of 10 mg/mL in methylene chloride or other suitable solvent. When 10 µL of this solution is spiked into a 1-mL extract, the concentration of the internal standards will be 100 µg/mL. A lower concentration appropriate to the response of the GC/MS instrument or for SIM may be used, if desired. Store per section 6.7.3.
6.9.3 To assure accurate analyte identification, particularly when SIM is used, it may be advantageous to include more internal standards than those suggested in section 6.9.1. An analyte will be located most accurately if its retention time relative to an internal standard is in the range of 0.8 to 1.2.
6.10 DFTPP standard - Prepare a solution of DFTPP in methanol or other suitable solvent such that 50 ng or less will be injected (see section 13.2). An alternative concentration may be used to compensate for specific injection volumes or to assure that the operating range of the instrument is not exceeded, so long as the total injected is 50 ng or less. Include benzidine and pentachlorophenol in this solution such that ≤100 ng of benzidine and ≤50 ng of pentachlorophenol will be injected.
6.11 Quality control check sample concentrate - See section 8.2.1.
6.12 GPC calibration solution.
6.12.1 Prepare a methylene chloride solution to contain corn oil, bis(2-ethylhexyl) phthalate (BEHP), perylene, and sulfur at the concentrations in section 6.12.2, or at concentrations appropriate to the response of the detector.
Note:Sulfur does not readily dissolve in methylene chloride, but is soluble in warm corn oil. The following procedure is suggested for preparation of the solution.
6.12.2 Weigh 8 mg sulfur and 2.5 g corn oil into a 100-mL volumetric flask and warm to dissolve the sulfur. Separately weigh 100 mg BEHP, 20 mg pentachlorophenol, and 2 mg perylene and add to flask. Bring to volume with methylene chloride and mix thoroughly.
6.12.3 Store the solution in an amber glass bottle with a fluoropolymer-lined screw cap at 0-6 °C. Protect from light. Refrigeration may cause the corn oil to precipitate. Before use, allow the solution to stand at room temperature until the corn oil dissolves, or warm slightly to aid in dissolution. Replace the solution every year, or more frequently if the response of a component changes.
6.13 Sulfur removal - Copper foil or powder (bright, non-oxidized), or tetrabutylammonium sulfite (TBA sulfite).
6.13.1 Copper foil, or powder - Fisher, Alfa Aesar 42455-18, 625 mesh, or equivalent. Cut copper foil into approximately 1-cm squares. Copper must be activated before it may be used, as described below:
6.13.1.1 Place the quantity of copper needed for sulfur removal (section 11.2.1.3) in a ground-glass-stoppered Erlenmeyer flask or bottle. Cover the foil or powder with methanol.
6.13.1.2 Add HCl dropwise (0.5-1.0 mL) while swirling, until the copper brightens.
6.13.1.3 Pour off the methanol/HCl and rinse 3 times with reagent water to remove all traces of acid, then 3 times with acetone, then 3 times with hexane.
6.13.1.4 For copper foil, cover with hexane after the final rinse. Store in a stoppered flask under nitrogen until used. For the powder, dry on a rotary evaporator or under a stream of nitrogen. Store in a stoppered flask under nitrogen until used. Inspect the copper foil or powder before each use. It must have a bright, non-oxidized appearance to be effective. Copper foil or powder that has oxidized may be reactivated using the procedure described above.
6.13.2 Tetrabutylammonium sodium sulfite (TBA sodium sulfite).
6.13.2.1 Tetrabutylammonium hydrogen sulfate, [CH3(CH2)3]4NHSO4.
6.13.2.2 Sodium sulfite, Na2SO3.
6.13.2.3 Dissolve approximately 3 g tetrabutylammonium hydrogen sulfate in 100 mL of reagent water in an amber bottle with fluoropolymer-lined screw cap. Extract with three 20-mL portions of hexane and discard the hexane extracts.
6.13.2.4 Add 25 g sodium sulfite to produce a saturated solution. Store at room temperature. Replace after 1 month.
6.14 DDT and endrin decomposition (breakdown) solution - Prepare a solution containing endrin at a concentration of 1 µg/mL and 4,4′-DDT at a concentration of 2 µg/mL, in isooctane or hexane. A 1-µL injection of this standard will contain 1 nanogram (ng) of endrin and 2 ng of DDT. The concentration of the solution may be adjusted by the laboratory to accommodate other injection volumes such that the same masses of the two analytes are introduced into the instrument.
7. Calibration7.1 Establish operating conditions equivalent to those in the footnote to Table 4 or 5 for the base/neutral or acid fraction, respectively. If a combined base/neutral/acid fraction will be analyzed, use the conditions in the footnote to Table 4. Alternative temperature program and flow rate conditions may be used. It is necessary to calibrate the GC/MS for the analytes of interest (Section 1.3) only.
7.2 Internal standard calibration.
7.2.1 Prepare calibration standards for the analytes of interest and surrogates at a minimum of five concentration levels by adding appropriate volumes of one or more stock standards to volumetric flasks. One of the calibration standards should be at a concentration at or below the ML specified in Table 1, 2, or 3, or as specified by a regulatory/control authority or in a permit. The ML value may be rounded to a whole number that is more convenient for preparing the standard, but must not exceed the ML in Table 1, 2, or 3 for those analytes which list ML values. Alternatively, the laboratory may establish a laboratory ML for each analyte based on the concentration in a nominal whole-volume sample that is equivalent to the concentration of the lowest calibration standard in a series of standards produced in the laboratory or obtained from a commercial vendor. The laboratory's ML must not exceed the ML in Table 1, 2, or 3, and the resulting calibration must meet the acceptance criteria in Section 7.2.3, based on the RSD, RSE, or R 2. The concentrations of the other calibration standards should correspond to the expected range of concentrations found in real samples or should define the working range of the GC/MS system for full-scan and/or SIM operation, as appropriate. A minimum of six concentration levels is required for a second order, non-linear (e.g., quadratic; ax 2 + bx + c = 0) calibration (section 7.2.3). Calibrations higher than second order are not allowed. To each calibration standard or standard mixture, add a known constant volume of the internal standard solution (section 6.9), and dilute to volume with methylene chloride.
Note:The large number of analytes in Tables 1 through 3 may not be soluble or stable in a single solution; multiple solutions may be required if a large number of analytes are to be determined simultaneously.
7.2.1.1 Prior to analysis of the calibration standards, inject the DFTPP standard (Section 6.10) and adjust the scan rate of the mass spectrometer to produce a minimum of 5 mass spectra across the DFTPP GC peak. Adjust instrument conditions until the DFTPP criteria in Table 9A or 9B are met. Calculate peak tailing factors for benzidine and pentachlorophenol. Calculation of the tailing factor is illustrated in Figure 1. The tailing factor for benzidine and pentachlorophenol must be <2; otherwise, adjust instrument conditions and either replace the column or break off a short section of the front end of the column, and repeat the test. Once the scan conditions are established, they must be used for analyses of all standards, blanks, and samples.
Note:The DFTPP spectrum may be evaluated by summing the intensities of the m/z's across the GC peak, subtracting the background at each m/z in a region of the chromatogram within 20 scans of but not including any part of, the DFTPP peak. The DFTPP spectrum may also be evaluated by fitting a Gaussian to each m/z and using the intensity at the maximum for each Gaussian or by integrating the area at each m/z and using the integrated areas. Other means may be used for evaluation of the DFTPP spectrum so long as the spectrum is not distorted to meet the criteria in Table 9A or 9B.
7.2.1.2 Analyze the mid-point combined base/neutral and acid calibration standard and enter or review the retention time, relative retention time, mass spectrum, and quantitation m/z in the data system for each analyte of interest, surrogate, and internal standard. If additional analytes (Table 3) are to be quantified, include these analytes in the standard. The mass spectrum for each analyte must be comprised of a minimum of 2 m/z's (Tables 4 and 5); 3 to 5 m/z's assure more reliable analyte identification. Suggested quantitation m/z's are shown in Tables 4 and 5 as the primary m/z. If an interference occurs at the primary m/z, use one of the secondary m/z's or an alternate m/z. A single m/z only is required for quantitation.
7.2.1.3 For SIM operation, determine the analytes in each descriptor, the quantitation m/z for each analyte (the quantitation m/z can be the same as for full-scan operation; section 7.2.1.2), the dwell time on each m/z for each analyte, and the beginning and ending retention time for each descriptor. Analyze the verification standard in scan mode to verify m/z's and establish retention times for the analytes. There must be a minimum of two m/z's for each analyte to assure analyte identification. To maintain sensitivity, the number of m/z's in a descriptor should be limited. For example, for a descriptor with 10 m/z's and a chromatographic peak width of 5 sec, a dwell time of 100 ms at each m/z would result in a scan time of 1 second and provide 5 scans across the GC peak. The quantitation m/z will usually be the most intense peak in the mass spectrum. The quantitation m/z and dwell time may be optimized for each analyte. The acquisition table used for SIM must take into account the mass defect (usually less than 0.2 Dalton) that can occur at each m/z monitored. Refer to the footnotes to Table 4 or 5 for establishing operating conditions and to section 7.2.1.1 for establishing scan conditions.
7.2.1.4 For combined scan and SIM operation, set up the scan segments and descriptors to meet requirements in sections 7.2.1.1-7.2.1.3. Analyze unfamiliar samples in the scan mode to assure that the analytes of interest are determined.
7.2.2 Analyze each calibration standard according to section 12 and tabulate the area at the quantitation m/z against concentration for each analyte of interest, surrogate, and internal standard. If an interference is encountered, use a secondary m/z (Table 4 or 5) for quantitation. Calculate a response factor (RF) for each analyte of interest at each concentration using Equation 1.
 where: As
= Area of the characteristic m/z for the analyte of interest or
surrogate. Ais = Area of the characteristic m/z for the internal
standard. Cis = Concentration of the internal standard (µg/mL). Cs
= Concentration of the analyte of interest or surrogate (µg/mL).
where: As
= Area of the characteristic m/z for the analyte of interest or
surrogate. Ais = Area of the characteristic m/z for the internal
standard. Cis = Concentration of the internal standard (µg/mL). Cs
= Concentration of the analyte of interest or surrogate (µg/mL).
7.2. Calculate the mean (average) and relative standard deviation (RSD) of the responses factors. If the RSD is less than 35%, the RF can be assumed to be invariant and the average RF can be used for calculations. Alternatively, the results can be used to fit a linear or quadratic regression of response ratios, As/Ais, vs. concentration ratios Cs/Cis. If used, the regression must be weighted inversely proportional to concentration. The coefficient of determination (R 2; Reference 10) of the weighted regression must be greater than 0.920 (this value roughly corresponds to the RSD limit of 35%). Alternatively, the relative standard error (Reference 11) may be used as an acceptance criterion. As with the RSD, the RSE must be less than 35%. If an RSE less than 35% cannot be achieved for a quadratic regression, system performance is unacceptable and the system must be adjusted and re-calibrated.
Note:Using capillary columns and current instrumentation, it is quite likely that a laboratory can calibrate the target analytes in this method and achieve a linearity metric (either RSD or RSE) well below 35%. Therefore, laboratories are permitted to use more stringent acceptance criteria for calibration than described here, for example, to harmonize their application of this method with those from other sources.
7.3 Calibration verification - The RF or calibration curve must be verified immediately after calibration and at the beginning of each 12-hour shift, by analysis of a standard at or near the concentration of the mid-point calibration standard (section 7.2.1). The standard(s) must be obtained from a second manufacturer or a manufacturer's batch prepared independently from the batch used for calibration. Traceability must be to a national standard, when available. Include the surrogates (section 6.8) in this solution. It is necessary to verify calibration for the analytes of interest (section 1.3) only.
Note:The 12-hour shift begins after the DFTPP (section 13.1) and DDT/endrin tests (if DDT and endrin are to be determined), and after analysis of the calibration verification standard. The 12-hour shift ends 12 hours later. The DFTPP, DDT/endrin, and calibration verification tests are outside of the 12-hour shift.
7.3.1 Analyze the calibration verification standard(s) beginning in section 12. Calculate the percent recovery of each analyte. Compare the recoveries for the analytes of interest against the acceptance criteria for recovery (Q) in Table 6, and the recoveries for the surrogates against the acceptance criteria in Table 8. If recovery of the analytes of interest and surrogates meet acceptance criteria, system performance is acceptable and analysis of samples may continue. If any individual recovery is outside its limit, system performance is unacceptable for that analyte.
Note:The large number of analytes in Tables 6 and 8 present a substantial probability that one or more will fail acceptance criteria when all analytes are tested simultaneously.
7.3.2 When one or more analytes fail acceptance criteria, analyze a second aliquot of the calibration verification standard and compare ONLY those analytes that failed the first test (section 7.3.1) with their respective acceptance criteria. If these analytes now pass, system performance is acceptable and analysis of samples may continue. A repeat failure of any analyte that failed the first test, however, will confirm a general problem with the measurement system. If this occurs, repair the system (section 7.2.1.1) and repeat the test (section 7.3.1), or prepare a fresh calibration standard and repeat the test. If calibration cannot be verified after maintenance or injection of the fresh calibration standard, re-calibrate the instrument.
Note:If it is necessary to perform a repeat verification test frequently; i.e., perform two tests in order to pass, it may be prudent to perform two injections in succession and review the results, rather than perform one injection, review the results, then perform the second injection if results from the first injection fail. To maintain the validity of the test and re-test, system maintenance and/or adjustment is not permitted between the injections.
7.3.3 Many of the analytes in Table 3 do not have QC acceptance criteria in Table 6, and some of the surrogates in Table 8 do not have acceptance criteria. If calibration is to be verified and other QC tests are to be performed for these analytes, acceptance criteria must be developed and applied. EPA has provided guidance for development of QC acceptance criteria (References 12 and 13). Alternatively, analytes that do not have acceptance criteria in Table 6 or Table 8 may be based on laboratory control charts, or 60 to 140% may be used.
7.3.4 Internal standard responses - Verify that detector sensitivity has not changed by comparing the response of each internal standard in the calibration verification standard (section 7.3) to the response of the respective internal standard in the midpoint calibration standard (section 7.2.1). The peak areas or heights of the internal standards in the calibration verification standard must be within 50% to 200% (1/2 to 2x) of their respective peak areas or heights in the mid-point calibration standard. If not, repeat the calibration verification test using a fresh calibration verification standard (7.3), or perform and document system repair. Subsequent to repair, repeat the calibration verification test (section 7.3.1). If the responses are still not within 50% to 200%, re-calibrate the instrument (section 7.2.2) and repeat the calibration verification test.
8. Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality assurance program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and ongoing analysis of spiked samples and blanks to evaluate and document data quality (40 CFR 136.7). The laboratory must maintain records to document the quality of data generated. Results of ongoing performance tests are compared with established QC acceptance criteria to determine if the results of analyses meet performance requirements of this method. When results of spiked samples do not meet the QC acceptance criteria in this method, a quality control check sample (laboratory control sample; LCS) must be analyzed to confirm that the measurements were performed in an in-control mode of operation. A laboratory may develop its own performance criteria (as QC acceptance criteria), provided such criteria are as or more restrictive than the criteria in this method.
8.1.1 The laboratory must make an initial demonstration of capability (DOC) to generate acceptable precision and recovery with this method. This demonstration is detailed in Section 8.2.
8.1.2 In recognition of advances that are occurring in analytical technology, and to overcome matrix interferences, the laboratory is permitted certain options (section 1.6 and 40 CFR 136.6(b)) to improve separations or lower the costs of measurements. These options may include alternate extraction, concentration, and cleanup procedures (e.g., solid-phase extraction; rotary-evaporator concentration; column chromatography cleanup), changes in column and type of mass spectrometer (40 CFR 136.6(b)(4)(xvi)). Alternate determinative techniques, such as substitution of spectroscopic or immunoassay techniques, and changes that degrade method performance, are not allowed. If an analytical technique other than GC/MS is used, that technique must have a specificity equal to or greater than the specificity of GC/MS for the analytes of interest. The laboratory is also encouraged to participate in inter-comparison and performance evaluation studies (see section 8.10).
8.1.2.1 Each time a modification is made to this method, the laboratory is required to repeat the procedure in section 8.2. If the detection limit of the method will be affected by the change, the laboratory must demonstrate that the MDLs (40 CFR part 136, appendix B) are lower than one-third the regulatory compliance limit or the MDLs in this method, whichever are greater. If calibration will be affected by the change, the instrument must be recalibrated per section 7. Once the modification is demonstrated to produce results equivalent or superior to results produced by this method, that modification may be used routinely thereafter, so long as the other requirements in this method are met (e.g., matrix spike/matrix spike duplicate recovery and relative percent difference).
8.1.2.1.1 If SPE, or another allowed method modification, is to be applied to a specific discharge, the laboratory must prepare and analyze matrix spike/matrix spike duplicate (MS/MSD) samples (section 8.3) and LCS samples (section 8.4). The laboratory must include surrogates (section 8.7) in each of the samples. The MS/MSD and LCS samples must be fortified with the analytes of interest (Section 1.3). If the modification is for nationwide use, MS/MSD samples must be prepared from a minimum of nine different discharges (See section 8.1.2.1.2), and all QC acceptance criteria in this method must be met. This evaluation only needs to be performed once other than for the routine QC required by this method (for example it could be performed by the vendor of the SPE materials) but any laboratory using that specific material must have the results of the study available. This includes a full data package with the raw data that will allow an independent reviewer to verify each determination and calculation performed by the laboratory (see section 8.1.2.2.5, items (a)-(q)).
8.1.2.1.2 Sample matrices on which MS/MSD tests must be performed for nationwide use of an allowed modification:
(a) Effluent from a POTW.
(b) ASTM D5905 Standard Specification for Substitute Wastewater.
(c) Sewage sludge, if sewage sludge will be in the permit.
(d) ASTM D1141 Standard Specification for Substitute Ocean Water, if ocean water will be in the permit.
(e) Untreated and treated wastewaters up to a total of nine matrix types (see https://www.epa.gov/eg/industrial-effluent-guidelines for a list of industrial categories with existing effluent guidelines).
(i) At least one of the above wastewater matrix types must have at least one of the following characteristics:
(A) Total suspended solids greater than 40 mg/L.
(B) Total dissolved solids greater than 100 mg/L.
(C) Oil and grease greater than 20 mg/L.
(D) NaCl greater than 120 mg/L.
(E) CaCO3 greater than 140 mg/L.
(ii) Results of MS/MSD tests must meet QC acceptance criteria in Section 8.3.
(f) A proficiency testing (PT) sample from a recognized provider, in addition to tests of the nine matrices (section 8.1.2.1.1).
8.1.2.2 The laboratory is required to maintain records of modifications made to this method. These records include the following, at a minimum:
8.1.2.2.1 The names, titles, and business street addresses, telephone numbers, and email addresses, of the analyst(s) that performed the analyses and modification, and of the quality control officer that witnessed and will verify the analyses and modifications.
8.1.2.2.2 A list of analytes, by name and CAS Registry Number.
8.1.2.2.3 A narrative stating reason(s) for the modifications.
8.1.2.2.4 Results from all quality control (QC) tests comparing the modified method to this method, including:
(a) Calibration (section 7).
(b) Calibration verification (section 7).
(c) Initial demonstration of capability (section 8.2).
(d) Analysis of blanks (section 8.5).
(e) Matrix spike/matrix spike duplicate analysis (section 8.3).
(f) Laboratory control sample analysis (section 8.4).
8.1.2.2.5 Data that will allow an independent reviewer to validate each determination by tracing the instrument output (peak height, area, or other signal) to the final result. These data are to include:
(a) Sample numbers and other identifiers.
(b) Extraction dates.
(c) Analysis dates and times.
(d) Analysis sequence/run chronology.
(e) Sample weight or volume (ssection 10).
(f) Extract volume prior to each cleanup step (sections 10 and 11).
(g) Extract volume after each cleanup step (section 11).
(h) Final extract volume prior to injection (sections 10 and 12).
(i) Injection volume (section 12.2.3).
(j) Sample or extract dilution (section 12.2.3.2).
(k) Instrument and operating conditions.
(l) Column (dimensions, material, etc).
(m) Operating conditions (temperature program, flow rate, etc).
(n) Detector (type, operating conditions, etc).
(o) Chromatograms, mass spectra, and other recordings of raw data.
(p) Quantitation reports, data system outputs, and other data to link the raw data to the results reported.
(q) A written Standard Operating Procedure (SOP).
8.1.2.2.6 Each individual laboratory wishing to use a given modification must perform the start-up tests in section 8.1.2 (e.g., DOC, MDL), with the modification as an integral part of this method prior to applying the modification to specific discharges. Results of the DOC must meet the QC acceptance criteria in Table 6 for the analytes of interest (section 1.3), and the MDLs must be equal to or lower than the MDLs in Tables 1, 2, or 3 for the analytes of interest.
8.1.3 Before analyzing samples, the laboratory must analyze a blank to demonstrate that interferences from the analytical system, labware, and reagents, are under control. Each time a batch of samples is extracted or reagents are changed, a blank must be extracted and analyzed as a safeguard against laboratory contamination. Requirements for the blank are given in section 8.5.
8.1.4 The laboratory must, on an ongoing basis, spike and analyze to monitor and evaluate method and laboratory performance on the sample matrix. The procedure for spiking and analysis is given in section 8.3.
8.1.5 The laboratory must, on an ongoing basis, demonstrate through analysis of a quality control check sample (laboratory control sample, LCS; on-going precision and recovery sample, OPR) that the measurement system is in control. This procedure is given in section 8.4.
8.1.6 The laboratory must maintain performance records to document the quality of data that is generated. This procedure is given in section 8.9.
8.1.7 The large number of analytes tested in performance tests in this method present a substantial probability that one or more will fail acceptance criteria when many analytes are tested simultaneously, and a re-test is allowed if this situation should occur. If, however, continued re-testing results in further repeated failures, the laboratory must document and report the failures (e.g., as qualifiers on results), unless the failures are not required to be reported as determined by the regulatory/control authority. Results associated with a QC failure for an analyte regulated in a discharge cannot be used to demonstrate regulatory compliance. QC failures do not relieve a discharger or permittee of reporting timely results.
8.2 Initial demonstration of capability (DOC) - To establish the ability to generate acceptable recovery and precision, the laboratory must perform the DOC in sections 8.2.1 through 8.2.6 for the analytes of interest. The laboratory must also establish MDLs for the analytes of interest using the MDL procedure at 40 CFR part 136, appendix B. The laboratory's MDLs must be equal to or lower than those listed in Tables 1, 2, or 3 or lower than one third the regulatory compliance limit, whichever is greater. For MDLs not listed in Tables 4 and 5, the laboratory must determine the MDLs using the MDL procedure at 40 CFR part 136, appendix B under the same conditions used to determine the MDLs for the analytes listed in Tables 1, 2, and 3. All procedures used in the analysis, including cleanup procedures, must be included in the DOC.
8.2.1 For the DOC, a QC check sample concentrate (LCS concentrate) containing each analyte of interest (section 1.3) is prepared in a water-miscible solvent. The QC check sample concentrate must be prepared independently from those used for calibration, but may be from the same source as the second-source standard used for calibration verification (Section 7.3). The concentrate should produce concentrations of the analytes of interest in water at the mid-point of the calibration range, and may be at the same concentration as the LCS (section 8.4). Multiple solutions may be required.
Note:QC check sample concentrates are no longer available from EPA.
8.2.2 Using a pipet or micro-syringe, prepare four LCSs by adding an appropriate volume of the concentrate to each of four aliquots of reagent water, and mix well. The volume of reagent water must be the same as the volume that will be used for the sample, blank (section 8.5), and MS/MSD (section 8.3). A volume of 1-L and a concentration of 100 µg/L were used to develop the QC acceptance criteria in Table 6. Also add an aliquot of the surrogate spiking solution (section 6.8) to the reagent-water aliquots.
8.2.3 Extract and analyze the four LCSs according to the method beginning in Section 10.
8.2.4 Calculate the average percent recovery (X) and the standard deviation of the percent recovery (s) for each analyte using the four results.
8.2.5 For each analyte, compare s and (X) with the corresponding acceptance criteria for precision and recovery in Table 6. For analytes in Table 3 not listed in Table 6, DOC QC acceptance criteria must be developed by the laboratory. EPA has provided guidance for development of QC acceptance criteria (References 12 and 13). Alternatively, acceptance criteria for analytes not listed in Table 6 may be based on laboratory control charts. If s and (X) for all analytes of interest meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may begin. If any individual s exceeds the precision limit or any individual (X) falls outside the range for recovery, system performance is unacceptable for that analyte.
Note:The large number of analytes in Tables 1-3 present a substantial probability that one or more will fail at least one of the acceptance criteria when many or all analytes are determined simultaneously. Therefore, the analyst is permitted to conduct a “re-test” as described in section 8.2.6.
8.2.6 When one or more of the analytes tested fail at least one of the acceptance criteria, repeat the test for only the analytes that failed. If results for these analytes pass, system performance is acceptable and analysis of samples and blanks may proceed. If one or more of the analytes again fail, system performance is unacceptable for the analytes that failed the acceptance criteria. Correct the problem and repeat the test (section 8.2). See section 8.1.7 for disposition of repeated failures.
Note:To maintain the validity of the test and re-test, system maintenance and/or adjustment is not permitted between this pair of tests.
8.3 Matrix spike and matrix spike duplicate (MS/MSD) - The purpose of the MS/MSD requirement is to provide data that demonstrate the effectiveness of the method as applied to the samples in question by a given laboratory, and both the data user (discharger, permittee, regulated entity, regulatory/control authority, customer, other) and the laboratory share responsibility for provision of such data. The data user should identify the sample and the analytes of interest (section 1.3) to be spiked and provide sufficient sample volume to perform MS/MSD analyses. The laboratory must, on an ongoing basis, spike at least 5% of the samples in duplicate from each discharge being monitored to assess accuracy (recovery and precision). If direction cannot be obtained from the data user, the laboratory must spike at least one sample in duplicate per extraction batch of up to 20 samples with the analytes in Table 1. Spiked sample results should be reported only to the data user whose sample was spiked, or as requested or required by a regulatory/control authority, or in a permit.
8.3.1 If, as in compliance monitoring, the concentration of a specific analyte will be checked against a regulatory concentration limit, the concentration of the spike should be at that limit; otherwise, the concentration of the spike should be one to five times higher than the background concentration determined in section 8.3.2, at or near the midpoint of the calibration range, or at the concentration in the LCS (section 8.4) whichever concentration would be larger.
8.3.2 Analyze one sample aliquot to determine the background concentration (B) of the each analyte of interest. If necessary, prepare a new check sample concentrate (section 8.2.1) appropriate for the background concentration. Spike and analyze two additional sample aliquots, and determine the concentration after spiking (A1 and A2) of each analyte. Calculate the percent recoveries (P1 and P2) as 100 (A1 − B)/T and 100 (A2 − B)/T, where T is the known true value of the spike. Also calculate the relative percent difference (RPD) between the concentrations (A1 and A2) as 200 |A1 − A2|/(A1 + A2). If necessary, adjust the concentrations used to calculate the RPD to account for differences in the volumes of the spiked aliquots.
8.3.3 Compare the percent recoveries (P1 and P2) and the RPD for each analyte in the MS/MSD aliquots with the corresponding QC acceptance criteria in Table 6. A laboratory may develop and apply QC acceptance criteria more restrictive than the criteria in Table 6, if desired.
8.3.3.1 If any individual P falls outside the designated range for recovery in either aliquot, or the RPD limit is exceeded, the result for the analyte in the unspiked sample is suspect. See Section 8.1.7 for disposition of failures.
8.3.3.2 The acceptance criteria in Table 6 were calculated to include an allowance for error in measurement of both the background and spike concentrations, assuming a spike to background ratio of 5:1. This error will be accounted for to the extent that the spike to background ratio approaches 5:1 (Reference 14) and is applied to spike concentrations of 100 µg/L and higher. If spiking is performed at a concentration lower than 100 µg/L, the laboratory must use the QC acceptance criteria in Table 6, the optional QC acceptance criteria calculated for the specific spike concentration in Table 7, or optional in-house criteria (section 8.3.4). To use the acceptance criteria in Table 7: (1) Calculate recovery (X′) using the equation in Table 7, substituting the spike concentration (T) for C; (2) Calculate overall precision (S′) using the equation in Table 7, substituting X′ for X; (3) Calculate the range for recovery at the spike concentration as (100 X′/T) ± 2.44(100 S′/T)% (Reference 14). For analytes in Table 3 not listed in Table 6, QC acceptance criteria must be developed by the laboratory. EPA has provided guidance for development of QC acceptance criteria (References 12 and 13). Alternatively, acceptance criteria may be based on laboratory control charts.
8.3.4 After analysis of a minimum of 20 MS/MSD samples for each target analyte and surrogate, and if the laboratory chooses to develop and apply the optional in-house QC limits (Section 8.3.3), the laboratory should calculate and apply the optional in-house QC limits for recovery and RPD of future MS/MSD samples (Section 8.3). The QC limits for recovery are calculated as the mean observed recovery ±3 standard deviations, and the upper QC limit for RPD is calculated as the mean RPD plus 3 standard deviations of the RPDs. The in-house QC limits must be updated at least every two years and re-established after any major change in the analytical instrumentation or process. If in-house QC limits are developed, at least 80% of the analytes tested in the MS/MSD must have in-house QC acceptance criteria that are tighter than those in Table 6, and the remaining analytes (those other than the analytes included in the 80%) must meet the acceptance criteria in Table 6. If an in-house QC limit for the RPD is greater than the limit in Table 6, then the limit in Table 6 must be used. Similarly, if an in-house lower limit for recovery is below the lower limit in Table 6, then the lower limit in Table 6 must be used, and if an in-house upper limit for recovery is above the upper limit in Table 6, then the upper limit in Table 6 must be used.
8.4 Laboratory control sample (LCS) - A QC check sample (laboratory control sample, LCS; on-going precision and recovery sample, OPR) containing each analyte of interest (Section 1.3) and surrogate must be prepared and analyzed with each extraction batch of up to 20 samples to demonstrate acceptable recovery of the analytes of interest from a clean sample matrix.
8.4.1 Prepare the LCS by adding QC check sample concentrate (section 8.2.1) to reagent water. Include all analytes of interest (section 1.3) in the LCS. The LCS may be the same sample prepared for the DOC (section 8.2.1). The volume of reagent water must be the same as the volume used for the sample, blank (section 8.5), and MS/MSD (Section 8.3). Also add an aliquot of the surrogate spiking solution (section 6.8). The concentration of the analytes in reagent water should be the same as the concentration in the DOC (section 8.2.2).
8.4.2 Analyze the LCS prior to analysis of field samples in the extraction batch. Determine the concentration (A) of each analyte. Calculate the percent recovery (PS) as 100 (A/T)%, where T is the true value of the concentration in the LCS.
8.4.3 Compare the percent recovery (PS) for each analyte with its corresponding QC acceptance criterion in Table 6. For analytes of interest in Table 3 not listed in Table 6, use the QC acceptance criteria developed for the LCS (section 8.4.5), or limits based on laboratory control charts. If the recoveries for all analytes of interest fall within their respective QC acceptance criteria, analysis of blanks and field samples may proceed. If any individual PS falls outside the range, proceed according to section 8.4.4.
Note:The large number of analytes in Tables 1-3 present a substantial probability that one or more will fail the acceptance criteria when all analytes are tested simultaneously. Because a re-test is allowed in event of failure (sections 8.1.7 and 8.4.3), it may be prudent to extract and analyze two LCSs together and evaluate results of the second analysis against the QC acceptance criteria only if an analyte fails the first test.
8.4.4 Repeat the test only for those analytes that failed to meet the acceptance criteria (PS). If these analytes now pass, system performance is acceptable and analysis of blanks and samples may proceed. Repeated failure, however, will confirm a general problem with the measurement system. If this occurs, repeat the test using a fresh LCS (section 8.2.2) or an LCS prepared with a fresh QC check sample concentrate (section 8.2.1), or perform and document system repair. Subsequent to analysis of the LCS prepared with a fresh sample concentrate, or to system repair, repeat the LCS test (section 8.4). If failure of the LCS indicates a systemic problem with samples in the batch, re-extract and re-analyze the samples in the batch. See section 8.1.7 for disposition of repeated failures.
Note:To maintain the validity of the test and re-test, system maintenance and/or adjustment is not permitted between the pair of tests.
8.4.5 After analysis of 20 LCS samples, and if the laboratory chooses to develop and apply in-house QC limits, the laboratory should calculate and apply in-house QC limits for recovery to future LCS samples (section 8.4). Limits for recovery in the LCS should be calculated as the mean recovery ±3 standard deviations. A minimum of 80% of the analytes tested for in the LCS must have QC acceptance criteria tighter than those in Table 6, and the remaining analytes (those other than the analytes included in the 80%) must meet the acceptance criteria in Table 6. If an in-house lower limit for recovery is lower than the lower limit in Table 6, the lower limit in Table 6 must be used, and if an in-house upper limit for recovery is higher than the upper limit in Table 6, the upper limit in Table 6 must be used. Many of the analytes and surrogates do not contain acceptance criteria. The laboratory should use 60-140% as interim acceptance criteria for recoveries of spiked analytes and surrogates that do not have recovery limits specified in Table 8, and at least 80% of the surrogates must meet the 60-140% interim criteria until in-house LCS and surrogate limits are developed. Alternatively, acceptance criteria for analytes that do not have recovery limits in Table 6 may be based on laboratory control charts. In-house QC acceptance criteria must be updated at least every two years.
8.5 Blank - A blank must be extracted and analyzed with each extraction batch to demonstrate that the reagents and equipment used for preparation and analysis are free from contamination.
8.5.1 Spike the surrogates into the blank. Extract and concentrate the blank using the same procedures and reagents used for the samples, LCS, and MS/MSD in the batch. Analyze the blank immediately after analysis of the LCS (section 8.4) and prior to analysis of the MS/MSD and samples to demonstrate freedom from contamination.
8.5.2 If an analyte of interest is found in the blank: At a concentration greater than the MDL for the analyte, at a concentration greater than one-third the regulatory compliance limit, or at a concentration greater than one-tenth the concentration in a sample in the extraction batch, whichever is greater, analysis of samples must be halted, and the problem corrected. If the contamination is traceable to the extraction batch, samples affected by the blank must be re-extracted and the extracts re-analyzed. If, however, continued re-testing results in repeated blank contamination, the laboratory must document and report the failures (e.g., as qualifiers on results), unless the failures are not required to be reported as determined by the regulatory/control authority. Results associated with blank contamination for an analyte regulated in a discharge cannot be used to demonstrate regulatory compliance. QC failures do not relieve a discharger or permittee of reporting timely results.
8.6 Internal standards responses.
8.6.1 Calibration verification - The responses (GC peak heights or areas) of the internal standards in the calibration verification must be within 50% to 200% (1/2 to 2x) of their respective responses in the mid-point calibration standard. If they are not, repeat the calibration verification (Section 7.4) test or perform and document system repair. Subsequent to repair, repeat the calibration verification. If the responses are still not within 50% to 200%, re-calibrate the instrument (Section 7) and repeat the calibration verification test.
8.6.2 Samples, blanks, LCSs, and MS/MSDs - The responses (GC peak heights or areas) of each internal standard in each sample, blank, and MS/MSD must be within 50% to 200% (1/2 to 2x) of its respective response in the LCS for the extraction batch. If, as a group, all internal standards are not within this range, perform and document system repair, repeat the calibration verification (section 8.4), and re-analyze the affected samples. If a single internal standard is not within the 50% to 200% range, use an alternate internal standard for quantitation of the analyte referenced to the affected internal standard. It may be necessary to use the data system to calculate a new response factor from calibration data for the alternate internal standard/analyte pair. If an internal standard fails the 50-200% criteria and no analytes are detected in the sample, ignore the failure or report it if required by the regulatory/control authority.
8.7 Surrogate recoveries - The laboratory must evaluate surrogate recovery data in each sample against its in-house surrogate recovery limits. The laboratory may use 60-140% as interim acceptance criteria for recoveries for surrogates not listed in Table 8. At least 80% of the surrogates must meet the 60-140% interim criteria until in-house limits are developed. Alternatively, surrogate recovery limits may be developed from laboratory control charts, but such limits must be at least as restrictive as those in Table 8. Spike the surrogates into all samples, blanks, LCSs, and MS/MSDs. Compare surrogate recoveries against the QC acceptance criteria in Table 8 and/or those developed in section 7.3.3 or 8.4.5. If any recovery fails its criteria, attempt to find and correct the cause of the failure. See section 8.1.7 for disposition of failures.
8.8 DDT and endrin decomposition (breakdown) - If DDT and/or endrin are to be analyzed using this method, the DDT/endrin decomposition test in section 13.8 must be performed to reliably quantify these two pesticides.
8.9 As part of the QC program for the laboratory, control charts or statements of accuracy for wastewater samples must be assessed and records maintained (40 CFR 136.7(c)(1)(viii)). After analysis of five or more spiked wastewater samples as in section 8.3, calculate the average percent recovery (PX) and the standard deviation of the percent recovery (sp). Express the accuracy assessment as a percent interval from PX −2sp to PX +2sp. For example, if PX = 90% and sp = 10%, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each analyte on a regular basis (e.g., after each 5-10 new accuracy measurements). If desired, statements of accuracy for laboratory performance, independent of performance on samples, may be developed using LCSs.
8.10 It is recommended that the laboratory adopt additional quality assurance practices for use with this method. The specific practices that are most productive depend upon the needs of the laboratory and the nature of the samples. Field duplicates may be analyzed to assess the precision of environmental measurements. Whenever possible, the laboratory should analyze standard reference materials and participate in relevant performance evaluation studies.
9. Sample Collection, Preservation, and Handling9.1 Collect samples as grab samples in amber or clear glass bottles, or in refrigerated bottles using automatic sampling equipment. If clear glass is used, protect samples from light. Collect 1-L of ambient waters, effluents, and other aqueous samples. If the sensitivity of the analytical system is sufficient, a smaller volume (e.g., 250 mL), but no less than 100 mL, may be used. Conventional sampling practices (Reference 15) should be followed, except that the bottle must not be pre-rinsed with sample before collection. Automatic sampling equipment must be as free as possible of polyvinyl chloride or other tubing or other potential sources of contamination. If needed, collect additional sample(s) for the MS/MSD (section 8.3).
9.2 Ice or refrigerate samples at ≤6 °C from the time of collection until extraction, but do not freeze. If residual chlorine is present, add 80 mg of sodium thiosulfate per liter of sample and mix well. Any method suitable for field use may be employed to test for residual chlorine (Reference 16). Add more sodium sulfate if 80 mg/L is insufficient but do not add excess sodium thiosulfate. If sodium thiosulfate interferes in the determination of the analytes, an alternate preservative (e.g., ascorbic acid or sodium sulfite) may be used. If preservative has been added, shake the sample vigorously for one minute. Maintain the hermetic seal on the sample bottle until time of analysis.
9.3 All samples must be extracted within 7 days of collection and sample extracts must be analyzed within 40 days of extraction.
10. Extraction10.1 This section contains procedures for separatory funnel liquid-liquid extraction (SFLLE) and continuous liquid-liquid extraction (CLLE). SFLLE is faster, but may not be as effective as CLLE for recovery of polar analytes such as phenol. SFLLE is labor intensive and may result in formation of emulsions that are difficult to break. CLLE is less labor intensive, avoids emulsion formation, but requires more time (18-24 hours) and more hood space, and may require more solvent. The procedures assume base-neutral extraction followed by acid extraction. For some matrices and analytes of interest, improved results may be obtained by acid-neutral extraction followed by base extraction. A single acid or base extraction may also be performed. If an extraction scheme alternate to base-neutral followed by acid extraction is used, all QC tests must be performed and all QC acceptance criteria must be met with that extraction scheme as an integral part of this method. Solid-phase extraction (SPE) may be used provided requirements in section 8.1.2 are met.
10.2 Separatory funnel liquid-liquid extraction (SFLLE) and extract concentration.
10.2.1 The SFLLE procedure below assumes a sample volume of 1 L. When a different sample volume is extracted, adjust the volume of methylene chloride accordingly.
10.2.2 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Pour the entire sample into the separatory funnel. Pipet the surrogate standard spiking solution (section 6.8) into the separatory funnel. If the sample will be used for the LCS or MS or MSD, pipet the appropriate check sample concentrate (section 8.2.1 or 8.3.2) into the separatory funnel. Mix well. Check the pH of the sample with wide-range pH paper and adjust to pH 11-13 with sodium hydroxide solution.
10.2.3 Add 60 mL of methylene chloride to the sample bottle, seal, and shake for approximately 30 seconds to rinse the inner surface. Transfer the solvent to the separatory funnel and extract the sample by shaking the funnel for two minutes with periodic venting to release excess pressure. Allow the organic layer to separate from the water phase for a minimum of 10 minutes. If the emulsion interface between layers is more than one-third the volume of the solvent layer, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration of the emulsion through glass wool or phase-separation paper, salting, centrifugation, or other physical methods. Collect the methylene chloride extract in a flask. If the emulsion cannot be broken (recovery of <80% of the methylene chloride), transfer the sample, solvent, and emulsion into a continuous extractor and proceed as described in section 10.3.
10.2.4 Add a second 60-mL volume of methylene chloride to the sample bottle and repeat the extraction procedure a second time, combining the extracts in the Erlenmeyer flask. Perform a third extraction in the same manner.
10.2.5 Adjust the pH of the aqueous phase to less than 2 using sulfuric acid. Serially extract the acidified aqueous phase three times with 60 mL aliquots of methylene chloride. Collect and combine the extracts in a flask in the same manner as the base/neutral extracts.
Note:Base/neutral and acid extracts may be combined for concentration and analysis provided all QC tests are performed and all QC acceptance criteria met for the analytes of interest with the combined extract as an integral part of this method, and provided that the analytes of interest are as reliably identified and quantified as when the extracts are analyzed separately. If doubt exists as to whether identification and quantitation will be affected by use of a combined extract, the fractions must be analyzed separately.
10.2.6 For each fraction or the combined fractions, assemble a Kuderna-Danish (K-D) concentrator by attaching a 10-mL concentrator tube to a 500-mL evaporative flask. Other concentration devices or techniques may be used in place of the K-D concentrator so long as the requirements in section 8.2 are met.
10.2.7 For each fraction or the combined fractions, pour the extract through a solvent-rinsed drying column containing about 10 cm of anhydrous sodium sulfate, and collect the extract in the K-D concentrator. Rinse the Erlenmeyer flask and column with 20-30 mL of methylene chloride to complete the quantitative transfer.
10.2.8 Add one or two clean boiling chips and attach a three-ball Snyder column to the evaporative flask for each fraction (section 10.2.7). Pre-wet the Snyder column by adding about 1 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60-65 °C) so that the concentrator tube is partially immersed in the hot water, and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15-20 minutes. At the proper rate of distillation, the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches 1 mL or other determined amount, remove the K-D apparatus from the water bath and allow to drain and cool for at least 10 minutes. Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1-2 mL of methylene chloride. A 5-mL syringe is recommended for this operation. If the sample will be cleaned up, reserve the K-D apparatus for concentration of the cleaned up extract. Adjust the volume to 5 mL with methylene chloride and proceed to section 11 for cleanup; otherwise, further concentrate the extract for GC/MS analysis per section 10.2.9 or 10.2.10.
10.2.9 Micro Kuderna-Danish concentration - Add another one or two clean boiling chips to the concentrator tube for each fraction and attach a two-ball micro-Snyder column. Pre-wet the Snyder column by adding about 0.5 mL of methylene chloride to the top. Place the K-D apparatus on a hot water bath (60-65 °C) so that the concentrator tube is partially immersed in hot water. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 5-10 minutes. At the proper rate of distillation the balls of the column will actively chatter but the chambers will not flood with condensed solvent. When the apparent volume of liquid reaches about 1 mL or other determined amount, remove the K-D apparatus from the water bath and allow it to drain and cool for at least 10 minutes. Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with approximately 0.2 mL of or methylene chloride. Adjust the final volume to 1.0 mL or a volume appropriate to the sensitivity desired (e.g., to meet lower MDLs or for selected ion monitoring). Record the volume, stopper the concentrator tube and store refrigerated if further processing will not be performed immediately. If the extracts will be stored longer than two days, they should be transferred to fluoropolymer-lined screw-cap vials and labeled base/neutral or acid fraction as appropriate. Mark the level of the extract on the vial so that solvent loss can be detected.
10.2.10 Nitrogen evaporation and solvent exchange - Extracts may be concentrated for analysis using nitrogen evaporation in place of micro K-D concentration (section 10.2.9). Extracts that have been cleaned up using sulfur removal (section 11.2) and are ready for analysis are exchanged into methylene chloride.
10.2.10.1 Transfer the vial containing the sample extract to the nitrogen evaporation (blowdown) device (section 5.8). Lower the vial into the water bath and begin concentrating. If the more volatile analytes (section 1.2) are to be concentrated, use room temperature for concentration; otherwise, a slightly elevated (e.g., 30-45 °C) may be used. During the solvent evaporation process, keep the solvent level below the water level of the bath and do not allow the extract to become dry. Adjust the flow of nitrogen so that the surface of the solvent is just visibly disturbed. A large vortex in the solvent may cause analyte loss.
10.2.10.2 Extracts to be solvent exchanged - When the volume of the liquid is approximately 200 µL, add 2 to 3 mL of methylene chloride and continue concentrating to approximately 100 µL. Repeat the addition of solvent and concentrate once more. Adjust the final extract volume to be consistent with the volume extracted and the sensitivity desired.
10.2.10.3 For extracts that have been cleaned up by GPC and that are to be concentrated to a nominal volume of 1 mL, adjust the final volume to compensate the GPC loss. For a 50% GPC loss, concentrate the extract to 1/2000 of the volume extracted. For example, if the volume extracted is 950 mL, adjust the final volume to 0.48 mL. For extracts that have not been cleaned up by GPC and are to be concentrated to a nominal volume of 1.0 mL, adjust the final extract volume to 1/1000 of the volume extracted. For example, if the volume extracted is 950 mL, adjust the final extract volume to 0.95 mL. Alternative means of compensating the loss during GPC are acceptable so long as they produce results as accurate as results produced using the procedure detailed in this Section. An alternative final volume may be used, if desired, and the calculations adjusted accordingly.
Note:The difference in the volume fraction for an extract cleaned up by GPC accounts for the loss in GPC cleanup. Also, by preserving the ratio between the volume extracted and the final extract volume, the concentrations and detection limits do not need to be adjusted for differences in the volume extracted and the extract volume.
10.2.11 Transfer the concentrated extract to a vial with fluoropolymer-lined cap. Seal the vial and label with the sample number. Store in the dark at room temperature until ready for GC analysis. If GC analysis will not be performed on the same day, store the vial in the dark at ≤6 °C. Analyze the extract by GC/MS per the procedure in section 12.
10.2.12 Determine the original sample volume by refilling the sample bottle to the mark and transferring the liquid to an appropriately sized graduated cylinder. For sample volumes on the order of 1000 mL, record the sample volume to the nearest 10 mL; for sample volumes on the order of 100 mL, record the volume to the nearest 1 mL. Sample volumes may also be determined by weighing the container before and after filling to the mark with water.
10.3 Continuous liquid/liquid extraction (CLLE).
Note:With CLLE, phenol, 2,4-dimethyl phenol, and some other analytes may be preferentially extracted into the base-neutral fraction. Determine an analyte in the fraction in which it is identified and quantified most reliably. Also, the short-chain phthalate esters (e.g., dimethyl phthalate, diethyl phthalate) and some other compounds may hydrolyze during prolonged exposure to basic conditions required for continuous extraction, resulting in low recovery of these analytes. When these analytes are of interest, their recovery may be improved by performing the acid extraction first.
10.3.1 Use CLLE when experience with a sample from a given source indicates an emulsion problem, or when an emulsion is encountered during SFLLE. CLLE may be used for all samples, if desired.
10.3.2 Mark the water meniscus on the side of the sample bottle for later determination of sample volume. Check the pH of the sample with wide-range pH paper and adjust to pH 11-13 with sodium hydroxide solution. Transfer the sample to the continuous extractor. Pipet surrogate standard spiking solution (section 6.8) into the sample. If the sample will be used for the LCS or MS or MSD, pipet the appropriate check sample concentrate (section 8.2.1 or 8.3.2) into the extractor. Mix well. Add 60 mL of methylene chloride to the sample bottle, seal, and shake for 30 seconds to rinse the inner surface. Transfer the solvent to the extractor.
10.3.3 Repeat the sample bottle rinse with an additional 50-100 mL portion of methylene chloride and add the rinse to the extractor.
10.3.4 Add a suitable volume of methylene chloride to the distilling flask (generally 200-500 mL), add sufficient reagent water to ensure proper operation, and extract for 18-24 hours. A shorter or longer extraction time may be used if all QC acceptance criteria are met. Test and, if necessary, adjust the pH of the water during the second or third hour of the extraction. After extraction, allow the apparatus to cool, then detach the distilling flask. Dry, concentrate, and seal the extract per sections 10.2.6 through 10.2.11. See the note at section 10.2.5 regarding combining extracts of the base/neutral and acid fractions.
10.3.5 Charge the distilling flask with methylene chloride and attach it to the continuous extractor. Carefully, while stirring, adjust the pH of the aqueous phase to less than 2 using sulfuric acid. Extract for 18-24 hours. A shorter or longer extraction time may be used if all QC acceptance criteria are met. Test and, if necessary, adjust the pH of the water during the second or third hour of the extraction. After extraction, allow the apparatus to cool, then detach the distilling flask. Dry, concentrate, and seal the extract per sections 10.2.6 through 10.2.11. Determine the sample volume per section 10.2.12.
11. Extract Cleanup Note:Cleanup may not be necessary for relatively clean samples (e.g., treated effluents, groundwater, drinking water). If particular circumstances require the use of a cleanup procedure, the laboratory may use any or all of the procedures below or any other appropriate procedure. Before using a cleanup procedure, the laboratory must demonstrate that the requirements of section 8.1.2 can be met using the cleanup procedure as an integral part of this method.
11.1 Gel permeation chromatography (GPC).
11.1.1 Calibration.
11.1.1.1 Load the calibration solution (section 6.12) into the sample loop.
11.1.1.2 Inject the calibration solution and record the signal from the detector. The elution pattern will be corn oil, bis(2-ethylhexyl) phthalate, pentachlorophenol, perylene, and sulfur.
11.1.1.3 Set the “dump time” to allow >85% removal of the corn oil and >85% collection of the phthalate.
11.1.1.4 Set the “collect time” to the peak minimum between perylene and sulfur.
11.1.1.5 Verify calibration with the calibration solution after every 20 or fewer extracts. Calibration is verified if the recovery of the pentachlorophenol is greater than 85%. If calibration is not verified, recalibrate using the calibration solution, and re-extract and clean up the preceding extracts using the calibrated GPC system.
11.1.2 Extract cleanup - GPC requires that the column not be overloaded. The column specified in this method is designed to handle a maximum of 0.5 g of high molecular weight material in a 5-mL extract. If the extract is known or expected to contain more than 0.5 g, the extract is split into fractions for GPC and the fractions are combined after elution from the column. The solids content of the extract may be obtained gravimetrically by evaporating the solvent from a 50-µL aliquot.
11.1.2.1 Filter the extract or load through the filter holder to remove particulates. Load the extract into the sample loop. The maximum capacity of the column is 0.5-1.0 g. If necessary, split the extract into multiple aliquots to prevent column overload.
11.1.2.2 Elute the extract using the calibration data determined in Section 11.1.1. Collect the eluate in the K-D apparatus reserved in section 10.2.8.
11.1.3 Concentrate the cleaned up extract per sections 10.2.8 and 10.2.9 or 10.2.10.
11.1.4 Rinse the sample loading tube thoroughly with methylene chloride between extracts to prepare for the next sample.
11.1.5 If a particularly dirty extract is encountered, run a methylene chloride blank through the system to check for carry-over.
11.2 Sulfur removal.
Note:Separate procedures using copper or TBA sulfite are provided in this section for sulfur removal. They may be used separately or in combination, if desired.
11.2.1 Removal with copper (Reference 17).
Note:If an additional compound (Table 3) is to be determined; sulfur is to be removed; copper will be used for sulfur removal; and a sulfur matrix is known or suspected to be present, the laboratory must demonstrate that the additional compound can be successfully extracted and treated with copper in the sulfur matrix. Some of the additional compounds (Table 3) are known not to be amenable to sulfur removal with copper (e.g., Atrazine and Diazinon).
11.2.1.1 Quantitatively transfer the extract from section 10.2.8 to a 40- to 50-mL flask or bottle. If there is evidence of water in the concentrator tube after the transfer, rinse the tube with small portions of hexane:acetone (40:60) and add to the flask or bottle. Mark and set aside the concentrator tube for use in re-concentrating the extract.
11.2.1.2 Add 10-20 g of granular anhydrous sodium sulfate to the flask. Swirl to dry the extract.
11.2.1.3 Add activated copper (section 6.13.1.4) and allow to stand for 30 - 60 minutes, swirling occasionally. If the copper does not remain bright, add more and swirl occasionally for another 30-60 minutes.
11.2.1.4 After drying and sulfur removal, quantitatively transfer the extract to a nitrogen-evaporation vial or tube and proceed to section 10.2.10 for nitrogen evaporation and solvent exchange, taking care to leave the sodium sulfate and copper in the flask.
11.2.2 Removal with TBA sulfite.
11.2.2.1 Using small volumes of hexane, quantitatively transfer the extract to a 40- to 50-mL centrifuge tube with fluoropolymer-lined screw cap.
11.2.2.2 Add 1-2 mL of TBA sulfite reagent (section 6.13.2.4), 2-3 mL of 2-propanol, and approximately 0.7 g of sodium sulfite (section 6.13.2.2) crystals to the tube. Cap and shake for 1-2 minutes. If the sample is colorless or if the initial color is unchanged, and if clear crystals (precipitated sodium sulfite) are observed, sufficient sodium sulfite is present. If the precipitated sodium sulfite disappears, add more crystalline sodium sulfite in approximately 0.5 g portions until a solid residue remains after repeated shaking.
11.2.2.3 Add 5-10 mL of reagent water and shake for 1-2 minutes. Centrifuge to settle the solids.
11.2.2.4 Quantitatively transfer the hexane (top) layer through a small funnel containing a few grams of granular anhydrous sodium sulfate to a nitrogen-evaporation vial or tube and proceed to section 10.2.10 for nitrogen evaporation and solvent exchange.
12. Gas Chromatography/Mass Spectrometry12.1 Establish the operating conditions in Table 4 or 5 for analysis of a base/neutral or acid extract, respectively. For analysis of a combined extract (section 10.2.5, note), use the operating conditions in Table 4 MDLs and MLs for the analytes are given in Tables 1, 2, and 3. Retention times for many of the analytes are given in Tables 4 and 5. Examples of the separations achieved are shown in Figure 2 for the combined extract. Alternative columns or chromatographic conditions may be used if the requirements of section 8.2 are met. Verify system performance per section 13.
12.2 Analysis of a standard or extract.
12.2.1 Bring the standard or concentrated extract (section 10.2.9 or 10.2.11) to room temperature and verify that any precipitate has redissolved. Verify the level on the extract and bring to the mark with solvent if required.
12.2.2 Add the internal standard solution (section 6.9) to the extract. Mix thoroughly.
12.2.3 Inject an appropriate volume of the sample extract or standard solution using split, splitless, solvent purge, large-volume, or on-column injection. If the sample is injected manually the solvent-flush technique should be used. The injection volume depends upon the technique used and the ability to meet MDLs or reporting limits for regulatory compliance. Injected volumes must be the same for standards and sample extracts. Record the volume injected to two significant figures.
12.2.3.1 Start the GC column oven program upon injection. Start MS data collection after the solvent peak elutes. Stop data collection after benzo(ghi)perylene elutes for the base/neutral or combined fractions, or after pentachlorophenol elutes for the acid fraction. Return the column to the initial temperature for analysis of the next standard solution or extract.
12.2.3.2 If the concentration of any analyte of interest exceeds the calibration range, either extract and analyze a smaller sample volume, or dilute and analyze the diluted extract after bringing the concentrations of the internal standards to the levels in the undiluted extract.
12.2.4 Perform all qualitative and quantitative measurements as described in Sections 14 and 15. When standards and extracts are not being used for analyses, store them refrigerated at ≤6 °C protected from light in screw-cap vials equipped with un-pierced fluoropolymer-lined septa.
13. Performance Tests13.1 At the beginning of each 12-hour shift during which standards or extracts will be analyzed, perform the tests in sections 13.2-13.4 to verify system performance. If an extract is concentrated for greater sensitivity (e.g., by SIM), all tests must be performed at levels consistent with the reduced extract volume.
13.2 DFTPP - Inject the DFTPP standard (section 6.10) and verify that the criteria for DFTPP in section 7.2.1.1 and Table 9A (Reference 18) for a quadrupole MS, or Table 9B (Reference 19) for a time-of-flight MS, are met.
13.3 GC resolution - The resolution should be verified on the mid-point concentration of the initial calibration as well as the laboratory designated continuing calibration verification level if closely eluting isomers are to be reported (e.g., benzo(b)fluoranthene and benzo(k)fluoranthene). Sufficient gas chromatographic resolution is achieved if the height of the valley between two isomer peaks is less than 50% of the average of the two peak heights.
13.4 Calibration verification - Verify calibration per sections 7.3 and Table 6.
13.5 Peak tailing - Verify the tailing factor specifications are met per Section 7.2.1.1.
13.6 Laboratory control sample and blank - Analyze the extracts of the LCS and blank at the beginning of analyses of samples in the extraction batch (section 3.1). The LCS must meet the requirements in section 8.4, and the blank must meet the requirements in section 8.5 before sample extracts may be analyzed.
13.7 Analysis of DFTPP, the DDT/Endrin decomposition test (if used), the LCS, and the blank are outside of the 12-hour analysis shift (section 3.1). The total time for DFTPP, DDT/Endrin, the LCS, the blank, and the 12-hour shift must not exceed 15 hours.
13.8 Decomposition of DDT and endrin - If DDT and/or endrin are to be determined, this test must be performed prior to calibration verification (section 13.4). The QC acceptance criteria (section 13.8.3) must be met before analyzing samples for DDE and/or Endrin. DDT decomposes to DDE and DDD. Endrin decomposes to endrin aldehyde and endrin ketone.
13.8.1 Inject 1 µL of the DDT and endrin decomposition solution (section 6.14). As noted in section 6.14, other injection volumes may be used as long as the concentrations of DDT and endrin in the solution are adjusted to introduce the masses of the two analytes into the instrument that are listed in section 6.14.
13.8.2 Measure the areas of the peaks for DDT, DDE, DDD, Endrin, Endrin aldehyde, and Endrin ketone. Calculate the percent breakdown as shown in the equations below:

13.8.3 Both the % breakdown of DDT and of Endrin must be less than 20%, otherwise the system is not performing acceptably for DDT and endrin. In this case, repair the GC column system that failed and repeat the performance tests (sections 13.2 to 13.6) until the specification is met.
Note:DDT and endrin decomposition are usually caused by accumulation of particulates in the injector and in the front end of the column. Cleaning and silanizing the injection port liner, and breaking off a short section of the front end of the column will usually eliminate the decomposition problem. Either of these corrective actions may affect retention times, GC resolution, and calibration linearity.
14. Qualitative Identification14.1 Identification is accomplished by comparison of data from analysis of a sample or blank with data stored in the GC/MS data system (sections 5.6.5 and 7.2.1.2). Identification of an analyte is confirmed per sections 14.1.1 through 14.1.4.
14.1.1 The signals for the quantitation and secondary m/z's stored in the data system for each analyte of interest must be present and must maximize within the same two consecutive scans.
14.1.2 The retention time for the analyte should be within ± 10 seconds of the analyte in the calibration verification run at the beginning of the shift (section 7.3 or 13.4).
Note:Retention time windows other than ± 10 seconds may be appropriate depending on the performance of the gas chromatograph or observed retention time drifts due to certain types of matrix effects. Relative retention time (RRT) may be used as an alternative to absolute retention times if retention time drift is a concern. RRT is a unitless quantity (see Sec. 22.2), although some procedures refer to “RRT units” in providing the specification for the agreement between the RRT values in the sample and the calibration verification or other standard. When significant retention time drifts are observed, dilutions or spiked samples may help the analyst determine the effects of the matrix on elution of the target analytes and to assist in qualitative identification.
14.1.3 Either the background corrected EICP areas, or the corrected relative intensities of the mass spectral peaks at the GC peak maximum, must agree within 50% to 200% (1/2 to 2 times) for the quantitation and secondary m/z's in the reference mass spectrum stored in the data system (section 7.2.1.2), or from a reference library. For example, if a peak has an intensity of 20% relative to the base peak, the analyte is identified if the intensity of the peak in the sample is in the range of 10% to 40% of the base peak. If identification is ambiguous, an experienced spectrometrist (section 1.7) must determine the presence or absence of the compound.
14.2 Structural isomers that produce very similar mass spectra should be identified as individual isomers if they have sufficiently different gas chromatographic retention times. Sufficient gas chromatographic resolution is achieved if the height of the valley between two isomer peaks is less than 50% of the average of the two peak heights. Otherwise, structural isomers are identified as isomeric pairs.
15. Calculations15.1 When an analyte has been identified, quantitation of that analyte is based on the integrated abundance from the EICP of the primary characteristic m/z in Table 4 or 5. Calculate the concentration in the extract using the response factor (RF) determined in Section 7.2.2 and Equation 2. If the concentration of an analyte exceeds the calibration range, dilute the extract by the minimum amount to bring the concentration into the calibration range, and re-analyze the extract. Determine a dilution factor (DF) from the amount of the dilution. For example, if the extract is diluted by a factor of 2, DF = 2.
 where:
Cex = Concentration of the analyte in the extract, in µg/mL, and
the other terms are as defined in section 7.2.2.
where:
Cex = Concentration of the analyte in the extract, in µg/mL, and
the other terms are as defined in section 7.2.2.
Calculate the concentration of the analyte in the sample using the concentration in the extract, the extract volume, the sample volume, and the dilution factor, per Equation 3:
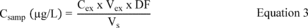 where:
Csamp = Concentration of the analyte in the sample Cex =
Concentration of the analyte in the extract, in µg/mL Vex = Volume
of extract (mL) Vs = Volume of sample (L) DF = Dilution factor
where:
Csamp = Concentration of the analyte in the sample Cex =
Concentration of the analyte in the extract, in µg/mL Vex = Volume
of extract (mL) Vs = Volume of sample (L) DF = Dilution factor
15.2 Reporting of results. As noted in section 1.4.1, EPA has promulgated this method at 40 CFR part 136 for use in wastewater compliance monitoring under the National Pollutant Discharge Elimination System (NPDES). The data reporting practices described here are focused on such monitoring needs and may not be relevant to other uses of the method.
15.2.1 Report results for wastewater samples in µg/L without correction for recovery. (Other units may be used if required by in a permit.) Report all QC data with the sample results.
15.2.2 Reporting level. Unless specified otherwise by a regulatory authority or in a discharge permit, results for analytes that meet the identification criteria are reported down to the concentration of the ML established by the laboratory through calibration of the instrument (see section 7.3.2 and the glossary for the derivation of the ML). EPA considers the terms “reporting limit,” “quantitation limit,” “limit of quantitation,” and “minimum level” to be synonymous.
15.2.2.1 Report a result for each analyte in each field sample or QC standard at or above the ML to 3 significant figures. Report a result for each analyte found in each field sample or QC standard below the ML as “ML” where ML is the concentration of the analyte at the ML, or as required by the regulatory/control authority or permit. Report a result for each analyte in a blank at or above the MDL to 2 significant figures. Report a result for each analyte found in a blank below the MDL as “MDL,” where MDL is the concentration of the analyte at the MDL, or as required by the regulatory/control authority or permit.
15.2.2.2 In addition to reporting results for samples and blanks separately, the concentration of each analyte in a blank associated with the sample may be subtracted from the result for that sample, but only if requested or required by a regulatory authority or in a permit. In this case, both the sample result and the blank results must be reported together.
15.2.2.3 Report a result for an analyte found in a sample or extract that has been diluted at the least dilute level at which the area at the quantitation m/z is within the calibration range (i.e., above the ML for the analyte) and the MS/MSD recovery and RPD are within their respective QC acceptance criteria (Table 6). This may require reporting results for some analytes from different analyses.
15.2.3 Results from tests performed with an analytical system that is not in control (i.e., that does not meet acceptance criteria for any QC test in this method) must be documented and reported (e.g., as a qualifier on results), unless the failure is not required to be reported as determined by the regulatory/control authority. Results associated with a QC failure cannot be used to demonstrate regulatory compliance. QC failures do not relieve a discharger or permittee of reporting timely results. If the holding time would be exceeded for a re-analysis of the sample, the regulatory/control authority should be consulted for disposition.
16. Method Performance16.1 The basic version of this method was tested by 15 laboratories using reagent water, drinking water, surface water, and industrial wastewaters spiked at six concentrations over the range 5-1300 µg/L (Reference 2). Single operator precision, overall precision, and method accuracy were found to be directly related to the concentration of the analyte and essentially independent of the sample matrix. Linear equations to describe these relationships are presented in Table 7.
16.2 As noted in section 1.1, this method was validated through an interlaboratory study in the early 1980s. However, the fundamental chemistry principles used in this method remain sound and continue to apply.
16.3 A chromatogram of the combined acid/base/neutral calibration standard is shown in Figure 2.
17. Pollution Prevention17.1 Pollution prevention encompasses any technique that reduces or eliminates the quantity or toxicity of waste at the point of generation. Many opportunities for pollution prevention exist in laboratory operations. EPA has established a preferred hierarchy of environmental management techniques that places pollution prevention as the management option of first choice. Whenever feasible, the laboratory should use pollution prevention techniques to address waste generation. When wastes cannot be reduced at the source, the Agency recommends recycling as the next best option.
17.2 The analytes in this method are used in extremely small amounts and pose little threat to the environment when managed properly. Standards should be prepared in volumes consistent with laboratory use to minimize the disposal of excess volumes of expired standards. This method utilizes significant quantities of methylene chloride. Laboratories are encouraged to recover and recycle this and other solvents during extract concentration.
17.3 For information about pollution prevention that may be applied to laboratories and research institutions, consult Less is Better: Laboratory Chemical Management for Waste Reduction, available from the American Chemical Society's Department of Governmental Relations and Science Policy, 1155 16th Street NW., Washington DC 20036, 202-872-4477.
18. Waste Management18.1 The laboratory is responsible for complying with all Federal, State, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions, and to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance is also required with any sewage discharge permits and regulations. An overview of requirements can be found in Environmental Management Guide for Small Laboratories (EPA 233-B-98-001).
18.2 Samples at pH <2, or pH >12, are hazardous and must be handled and disposed of as hazardous waste, or neutralized and disposed of in accordance with all federal, state, and local regulations. It is the laboratory's responsibility to comply with all federal, state, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions. The laboratory using this method has the responsibility to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance is also required with any sewage discharge permits and regulations. For further information on waste management, see “The Waste Management Manual for Laboratory Personnel,” also available from the American Chemical Society at the address in section 17.3.
18.3 Many analytes in this method decompose above 500 ºC. Low-level waste such as absorbent paper, tissues, and plastic gloves may be burned in an appropriate incinerator. Gross quantities of neat or highly concentrated solutions of toxic or hazardous chemicals should be packaged securely and disposed of through commercial or governmental channels that are capable of handling these types of wastes.
18.4 For further information on waste management, consult The Waste Management Manual for Laboratory Personnel and Less is Better-Laboratory Chemical Management for Waste Reduction, available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street NW., Washington, DC 20036, 202-872-4477.
19. References 1. “Sampling and Analysis Procedures for Screening of Industrial Effluents for Priority Pollutants,” U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268, March 1977, Revised April 1977. 2. “EPA Method Study 30, Method 625, Base/Neutrals, Acids, and Pesticides,” EPA 600/4-84-053, National Technical Information Service, PB84-206572, Springfield, Virginia 22161, June 1984. 3. 40 CFR part 136, appendix B. 4. Olynyk, P., Budde, W.L. and Eichelberger, J.W. “Method Detection Limit for Methods 624 and 625,” Unpublished report, May 14, 1980. 5. Annual Book of ASTM Standards, Volume 11.02, D3694-96, “Standard Practices for Preparation of Sample Containers and for Preservation of Organic Constituents,” American Society for Testing and Materials, Philadelphia. 6. Solutions to Analytical Chemistry Problems with Clean Water Act Methods, EPA 821-R-07-002, March 2007. 7. “Carcinogens-Working With Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977. 8. “OSHA Safety and Health Standards, General Industry,” (29 CFR part 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976). 9. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety, 7th Edition, 2003. 10. Johnson, R.A., and Wichern, D.W., “Applied Multivariate Statistical Analysis,” 3rd edition, Prentice Hall, Englewood Cliffs, NJ, 1992. 11. 40 CFR 136.6(b)(4)(x). 12. 40 CFR 136.6(b)(2)(i). 13. Protocol for EPA Approval of New Methods for Organic and Inorganic Analytes in Wastewater and Drinking Water (EPA-821-B-98-003) March 1999. 14. Provost, L.P. and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15, 58-63 (1983). (The value 2.44 used in the equation in section 8.3.3 is two times the value 1.22 derived in this report.) 15. ASTM Annual Book of Standards, Part 31, D3370-76. “Standard Practices for Sampling Water,” American Society for Testing and Materials, Philadelphia. 16. 40 CFR 136.3(a), Table IB, Chlorine - Total Residual. 17. “Manual of Analytical Methods for the Analysis of Pesticides in Human and Environmental Samples,” EPA-600/8-80-038, U.S. Environmental Protection Agency, Health Effects Research Laboratory, Research Triangle Park, North Carolina. 18. Eichelberger, J.W., Harris, L.E., and Budde, W.L. “Reference Compound to Calibrate Ion Abundance Measurement in Gas Chromatography-Mass Spectrometry,” Analytical Chemistry, 47, 995 (1975). 19. Letter of approval of acceptance criteria for DFTPP for time-of-flight mass spectrometers from William A. Telliard and Herb Brass of EPA to Jack Cochran of LECO Corporation, February 9, 2005. 20. TablesTable 1 - Non Pesticide/PCB Base/Neutral Extractables 1
| Analyte | CAS registry | MDL 4 (ug/L) |
ML 5 (ug/L) |
|---|---|---|---|
| Acenaphthene | 83-32-9 | 1.9 | 5.7 |
| Acenaphthylene | 208-96-8 | 3.5 | 10.5 |
| Anthracene | 120-12-7 | 1.9 | 5.7 |
| Benzidine 2 | 92-87-5 | 44 | 132 |
| Benzo(a)anthracene | 56-55-3 | 7.8 | 23.4 |
| Benzo(a)pyrene | 50-32-8 | 2.5 | 7.5 |
| Benzo(b)fluoranthene | 205-99-2 | 4.8 | 14.4 |
| Benzo(k)fluoranthene | 207-08-9 | 2.5 | 7.5 |
| Benzo(ghi)perylene | 191-24-2 | 4.1 | 12.3 |
| Benzyl butyl phthalate | 85-68-7 | 2.5 | 7.5 |
| bis(2-Chloroethoxy)methane | 111-91-1 | 5.3 | 15.9 |
| bis(2-Ethylhexyl)phthalate | 117-81-7 | 2.5 | 7.5 |
| bis(2-Chloroisopropyl) ether (2,2'-Oxybis[1-chloropropane]) | 108-60-1 | 5.7 | 17.1 |
| 4-Bromophenyl phenyl ether | 101-55-3 | 1.9 | 5.7 |
| 2-Chloronaphthalene | 91-58-7 | 1.9 | 5.7 |
| 4-Chlorophenyl phenyl ether | 7005-72-3 | 4.2 | 12.6 |
| Chrysene | 218-01-9 | 2.5 | 7.5 |
| Dibenz(a,h)anthracene | 53-70-3 | 2.5 | 7.5 |
| Di-n-butylphthalate | 84-74-2 | 2.5 | 7.5 |
| 3,3'-Dichlorobenzidine | 91-94-1 | 16.5 | 49.5 |
| Diethyl phthalate | 84-66-2 | 1.9 | 5.7 |
| Dimethyl phthalate | 131-11-3 | 1.6 | 4.8 |
| 2,4-Dinitrotoluene | 121-14-2 | 5.7 | 17.1 |
| 2,6-Dinitrotoluene | 606-20-2 | 1.9 | 5.7 |
| Di-n-octylphthalate | 117-84-0 | 2.5 | 7.5 |
| Fluoranthene | 206-44-0 | 2.2 | 6.6 |
| Fluorene | 86-73-7 | 1.9 | 5.7 |
| Hexachlorobenzene | 118-74-1 | 1.9 | 5.7 |
| Hexachlorobutadiene | 87-68-3 | 0.9 | 2.7 |
| Hexachloroethane | 67-72-1 | 1.6 | 4.8 |
| Indeno(1,2,3-cd)pyrene | 193-39-5 | 3.7 | 11.1 |
| Isophorone | 78-59-1 | 2.2 | 6.6 |
| Naphthalene | 91-20-3 | 1.6 | 4.8 |
| Nitrobenzene | 98-95-3 | 1.9 | 5.7 |
| N-Nitrosodi-n-propylamine 3 | 621-64-7 | - | - |
| Phenanthrene | 85-01-8 | 5.4 | 16.2 |
| Pyrene | 129-00-0 | 1.9 | 5.7 |
| 1,2,4-Trichlorobenzene | 120-82-1 | 1.9 | 5.7 |
1 All analytes in this table are Priority Pollutants (40 CFR part 423, appendix A).
2 Included for tailing factor testing.
3 See section 1.2.
4 MDL values from the 1984 promulgated version of Method 625.
5 ML = Minimum Level - see Glossary for definition and derivation.
Table 2 - Acid Extractables 1
| Analyte | CAS registry | MDL 3 (ug/L) |
ML 4 (ug/L) |
|---|---|---|---|
| 4-Chloro-3-methylphenol | 59-50-7 | 3.0 | 9.0 |
| 2-Chlorophenol | 95-57-8 | 3.3 | 9.9 |
| 2,4-Dichlorophenol | 120-83-2 | 2.7 | 8.1 |
| 2,4-Dimethylphenol | 105-67-9 | 2.7 | 8.1 |
| 2,4-Dinitrophenol | 51-28-5 | 42 | 126 |
| 2-Methyl-4,6-dinitrophenol | 534-52-1 | 24 | 72 |
| 2-Nitrophenol | 88-75-5 | 3.6 | 10.8 |
| 4-Nitrophenol | 100-02-7 | 2.4 | 7.2 |
| Pentachlorophenol 2 | 87-86-5 | 3.6 | 10.8 |
| Phenol | 108-95-2 | 1.5 | 4.5 |
| 2,4,6-Trichlorophenol | 88-06-2 | 2.7 | 8.1 |
1 All analytes in this table are Priority Pollutants (40 CFR part 423, appendix A).
2 See section 1.2; included for tailing factor testing.
3 MDL values from the 1984 promulgated version of Method 625.
4 ML = Minimum Level - see Glossary for definition and derivation.
Table 3 - Additional Extractable Analytes 1 2
| Analyte | CAS registry | MDL 7 (ug/L) |
ML 8 (ug/L) |
|---|---|---|---|
| Acetophenone | 98-86-2 | ||
| 2-Acetylaminofluorene | 53-96-3 | ||
| 1-Acetyl-2-thiourea | 591-08-2 | ||
| Alachlor | 15972-60-8 | ||
| Aldrin 3 | 309-00-2 | 1.9 | 5.7 |
| Ametryn | 834-12-8 | ||
| 2-Aminoanthraquinone | 117-79-3 | ||
| Aminoazobenzene | 60-09-3 | ||
| 4-Aminobiphenyl | 92-67-1 | ||
| 3-Amino-9-ethylcarbazole | 132-32-1 | ||
| Anilazine | 101-05-3 | ||
| Aniline | 62-53-3 | ||
| o-Anisidine | 90-04-0 | ||
| Aramite | 140-57-8 | ||
| Atraton | 1610-17-9 | ||
| Atrazine | 1912-24-9 | ||
| Azinphos-methyl | 86-50-0 | ||
| Barban | 101-27-9 | ||
| Benzanthrone | 82-05-3 | ||
| Benzenethiol | 108-98-5 | ||
| Benzoic acid | 65-85-0 | ||
| 2,3-Benzofluorene | 243-17-4 | ||
| p-Benzoquinone | 106-51-4 | ||
| Benzyl alcohol | 100-51-6 | ||
| alpha-BHC 3 4 | 319-84-6 | ||
| beta-BHC 3 | 319-85-7 | 3.1 | 9.3 |
| gamma-BHC (Lindane) 3 4 | 58-89-8 | 4.2 | 12.6 |
| delta-BHC 3 | 319-86-8 | ||
| Biphenyl | 92-52-4 | ||
| Bromacil | 314-40-9 | ||
| 2-Bromochlorobenzene | 694-80-4 | ||
| 3-Bromochlorobenzene | 108-39-2 | ||
| Bromoxynil | 1689-84-5 | ||
| Butachlor | 2318-4669 | ||
| Butylate | 2008-41-5 | ||
| n-C10 (n-decane) | 124-18-5 | ||
| n-C12 (n-undecane) | 112-40-2 | ||
| n-C14 (n-tetradecane) | 629-59-4 | ||
| n-C16 (n-hexadecane) | 544-76-3 | ||
| n-C18 (n-octadecane) | 593-45-3 | ||
| n-C20 (n-eicosane) | 112-95-8 | ||
| n-C22 (n-docosane) | 629-97-0 | ||
| n-C24 (n-tetracosane) | 646-31-1 | ||
| n-C26 (n-hexacosane) | 630-01-3 | ||
| n-C28 (n-octacosane) | 630-02-4 | ||
| n-C30 (n-triacontane) | 638-68-6 | ||
| Captafol | 2425-06-1 | ||
| Captan | 133-06-2 | ||
| Carbaryl | 63-25-2 | ||
| Carbazole | 86-74-8 | ||
| Carbofuran | 1563-66-2 | ||
| Carboxin | 5234-68 -4 | ||
| Carbophenothion | 786-19-6 | ||
| Chlordane 3 5 | 57-74-9 | ||
| bis(2-Chloroethyl) ether 3 4 | 111-44-4 | 5.7 | 17.1 |
| Chloroneb | 2675-77-6 | ||
| 4-Chloroaniline | 106-47-8 | ||
| Chlorobenzilate | 510-15-6 | ||
| Chlorfenvinphos | 470-90-6 | ||
| 4-Chloro-2-methylaniline | 95-69-2 | ||
| 3-(Chloromethyl)pyridine hydrochloride | 6959-48-4 | ||
| 4-Chloro-2-nitroaniline | 89-63-4 | ||
| Chlorpropham | 101-21-3 | ||
| Chlorothalonil | 1897-45-6 | ||
| 1-Chloronaphthalene | 90-13-1 | ||
| 3-Chloronitrobenzene | 121-73-3 | ||
| 4-Chloro-1,2-phenylenediamine | 95-83-0 | ||
| 4-Chloro-1,3-phenylenediamine | 5131-60-2 | ||
| 2-Chlorobiphenyl | 2051-60-7 | ||
| Chlorpyrifos | 2921-88-2 | ||
| Coumaphos | 56-72-4 | ||
| m + p-Cresol | 65794-96-9 | ||
| o-Cresol | 95-48-7 | ||
| p-Cresidine | 120-71-8 | ||
| Crotoxyphos | 7700-17-6 | ||
| 2-Cyclohexyl-4,6-dinitro-phenol | 131-89-5 | ||
| Cyanazine | 21725-46-2 | ||
| Cycloate | 1134-23-2 | ||
| p-Cymene | 99-87-6 | ||
| Dacthal (DCPA) | 1861-32-1 | ||
| 4,4'-DDD 3 | 72-54-8 | 2.8 | 8.4 |
| 4,4'-DDE 3 | 72-55-9 | 5.6 | 16.8 |
| 4,4'-DDT 3 | 50-29-3 | 4.7 | 14.1 |
| Demeton-O | 298-03-3 | ||
| Demeton-S | 126-75-0 | ||
| Diallate (cis or trans) | 2303-16-4 | ||
| 2,4-Diaminotoluene | 95-80-7 | ||
| Diazinon | 333-41-5 | ||
| Dibenz(a,j)acridine | 224-42-0 | ||
| Dibenzofuran | 132-64-9 | ||
| Dibenzo(a,e)pyrene | 192-65-4 | ||
| Dibenzothiophene | 132-65-0 | ||
| 1,2-Dibromo-3-chloropropane | 96-12-8 | ||
| 3,5-Dibromo-4-hydroxybenzonitrile | 1689-84-5 | ||
| 2,6-Di-tert-butyl-p-benzoquinone | 719-22-2 | ||
| Dichlone | 117-80-6 | ||
| 2,3-Dichloroaniline | 608-27-5 | ||
| 2,3-Dichlorobiphenyl | 16605-91-7 | ||
| 2,6-Dichloro-4-nitroaniline | 99-30-9 | ||
| 2,3-Dichloronitrobenzene | 3209-22-1 | ||
| 1,3-Dichloro-2-propanol | 96-23-1 | ||
| 2,6-Dichlorophenol | 120-83-2 | ||
| Dichlorvos | 62-73-7 | ||
| Dicrotophos | 141-66-2 | ||
| Dieldrin 3 | 60-57-1 | 2.5 | 7.5 |
| 1,2:3,4-Diepoxybutane | 1464-53-5 | ||
| Di(2-ethylhexyl) adipate | 103-23-1 | ||
| Diethylstilbestrol | 56-53-1 | ||
| Diethyl sulfate | 64-67-5 | ||
| Dilantin (5,5-Diphenylhydantoin) | 57-41-0 | ||
| Dimethoate | 60-51-5 | ||
| 3,3′-Dimethoxybenzidine | 119-90-4 | ||
| Dimethylaminoazobenzene | 60-11-7 | ||
| 7,12-Dimethylbenz(a)anthracene | 57-97-6 | ||
| 3,3′-Dimethylbenzidine | 119-93-7 | ||
| N,N-Dimethylformamide | 68-12-2 | ||
| 3,6-Dimethylphenathrene | 1576-67-6 | ||
| alpha, alpha-Dimethylphenethylamine | 122-09-8 | ||
| Dimethyl sulfone | 67-71-0 | ||
| 1,2-Dinitrobenzene | 528-29-0 | ||
| 1,3-Dinitrobenzene | 99-65-0 | ||
| 1,4-Dinitrobenzene | 100-25-4 | ||
| Dinocap | 39300-45-3 | ||
| Dinoseb | 88-85-7 | ||
| Diphenylamine | 122-39-4 | ||
| Diphenyl ether | 101-84-8 | ||
| 1,2-Diphenylhydrazine | 122-66-7 | ||
| Diphenamid | 957-51-7 | ||
| Diphenyldisulfide | 882-33-7 | ||
| Disulfoton | 298-04-4 | ||
| Disulfoton sulfoxide | 2497-07-6 | ||
| Disulfoton sulfone | 2497-06-5 | ||
| Endosulfan I 4 | 959-98-8 | ||
| Endosulfan II 3 4 | 33213-65-9 | ||
| Endosulfan sulfate 3 | 1031-07-8 | 5.6 | 16.8 |
| Endrin 3 4 | 72-20-8 | ||
| Endrin aldehyde 3 4 | 7421-93-4 | ||
| Endrin ketone 3 4 | 53494-70-5 | ||
| EPN | 2104-64-5 | ||
| EPTC | 759-94-4 | ||
| Ethion | 563-12-2 | ||
| Ethoprop | 13194-48-4 | ||
| Ethyl carbamate | 51-79-6 | ||
| Ethyl methanesulfonate | 65-50-0 | ||
| Ethylenethiourea | 96-45-7 | ||
| Etridiazole | 2593-15-9 | ||
| Ethynylestradiol-3-methyl ether | 72-33-3 | ||
| Famphur | 52-85-7 | ||
| Fenamiphos | 22224-92-6 | ||
| Fenarimol | 60168-88-9 | ||
| Fensulfothion | 115-90-2 | ||
| Fenthion | 55-38-9 | ||
| Fluchloralin | 33245-39-5 | ||
| Fluridone | 59756-60-4 | ||
| Heptachlor 3 | 76-44-8 | 1.9 | 5.7 |
| Heptachlor epoxide 3 | 1024-57-3 | 2.2 | 6.6 |
| 2,2′,3,3′,4,4′,6-Heptachlorobiphenyl | 52663-71-5 | ||
| 2,2′,4,4′,5′,6-Hexachlorobiphenyl | 60145-22-4 | ||
| Hexachlorocyclopentadiene 3 4 | 77-47-4 | ||
| Hexachlorophene | 70-30-4 | ||
| Hexachloropropene | 1888-71-7 | ||
| Hexamethylphosphoramide | 680-31-9 | ||
| Hexanoic acid | 142-62-1 | ||
| Hexazinone | 51235-04-2 | ||
| Hydroquinone | 123-31-9 | ||
| Isodrin | 465-73-6 | ||
| 2-Isopropylnaphthalene | 2027-17-0 | ||
| Isosafrole | 120-58-1 | ||
| Kepone | 143-50-0 | ||
| Leptophos | 21609-90-5 | ||
| Longifolene | 475-20-7 | ||
| Malachite green | 569-64-2 | ||
| Malathion | 121-75-5 | ||
| Maleic anhydride | 108-31-6 | ||
| Merphos | 150-50-5 | ||
| Mestranol | 72-33-3 | ||
| Methapyrilene | 91-80-5 | ||
| Methoxychlor | 72-43-5 | ||
| 2-Methylbenzothioazole | 120-75-2 | ||
| 3-Methylcholanthrene | 56-49-5 | ||
| 4,4′-Methylenebis(2-chloroaniline) | 101-14-4 | ||
| 4,4′-Methylenebis(N,N-dimethylaniline) | 101-61-1 | ||
| 4,5-Methylenephenanthrene | 203-64-5 | ||
| 1-Methylfluorene | 1730-37-6 | ||
| Methyl methanesulfonate | 66-27-3 | ||
| 2-Methylnaphthalene | 91-57-6 | ||
| Methylparaoxon | 950-35-6 | ||
| Methyl parathion | 298-00-0 | ||
| 1-Methylphenanthrene | 832-69-9 | ||
| 2-(Methylthio)benzothiazole | 615-22-5 | ||
| Metolachlor | 5218-45-2 | ||
| Metribuzin | 21087-64-9 | ||
| Mevinphos | 7786-34-7 | ||
| Mexacarbate | 315-18-4 | ||
| MGK 264 | 113-48-4 | ||
| Mirex | 2385-85-5 | ||
| Molinate | 2212-67-1 | ||
| Monocrotophos | 6923-22-4 | ||
| Naled | 300-76-5 | ||
| Napropamide | 15299-99-7 | ||
| 1,4-Naphthoquinone | 130-15-4 | ||
| 1-Naphthylamine | 134-32-7 | ||
| 2-Naphthylamine | 91-59-8 | ||
| 1,5-Naphthalenediamine | 2243-62-1 | ||
| Nicotine | 54-11-5 | ||
| 5-Nitroacenaphthene | 602-87-9 | ||
| 2-Nitroaniline | 88-74-4 | ||
| 3-Nitroaniline | 99-09-2 | ||
| 4-Nitroaniline | 100-01-6 | ||
| 5-Nitro-o-anisidine | 99-59-2 | ||
| 4-Nitrobiphenyl | 92-93-3 | ||
| Nitrofen | 1836-75-5 | ||
| 5-Nitro-o-toluidine | 99-55-8 | ||
| Nitroquinoline-1-oxide | 56-57-5 | ||
| N-Nitrosodi-n-butylamine 4 | 924-16-3 | ||
| N-Nitrosodiethylamine 4 | 55-18-5 | ||
| N-Nitrosodimethylamine 3 4 | 62-75-9 | ||
| N-Nitrosodiphenylamine 3 4 | 86-30-6 | ||
| N-Nitrosomethylethylamine 4 | 10595-95-6 | ||
| N-Nitrosomethylphenylamine 4 | 614-00-6 | ||
| N-Nitrosomorpholine 4 | 59-89-2 | ||
| N-Nitrosopiperidine 4 | 100-75-5 | ||
| N-Nitrosopyrrolidine 4 | 930-55-2 | ||
| trans-Nonachlor | 39765-80-5 | ||
| Norflurazon | 27314-13-2 | ||
| 2,2′,3,3′,4,5′,6,6′-Octachlorobiphenyl | 40186-71-8 | ||
| Octamethyl pyrophosphoramide | 152-16-9 | ||
| 4,4'-Oxydianiline | 101-80-4 | ||
| Parathion | 56-38-2 | ||
| PCB-1016 3 5 | 12674-11-2 | ||
| PCB-1221 3 5 | 11104-28-2 | 30 | 90 |
| PCB-1232 3 5 | 11141-16-5 | ||
| PCB-1242 3 5 | 53469-21-9 | ||
| PCB-1248 3 5 | 12672-29-6 | ||
| PCB-1254 3 5 | 11097-69-1 | 36 | 108 |
| PCB-1260 3 5 | 11098-82-5 | ||
| PCB-1268 3 5 | 11100-14-4 | ||
| Pebulate | 1114-71-2 | ||
| Pentachlorobenzene | 608-93-5 | ||
| Pentachloronitrobenzene | 82-68-8 | ||
| 2,2′,3,4′,6-Pentachlorobiphenyl | 68194-05-8 | ||
| Pentachloroethane | 76-01-7 | ||
| Pentamethylbenzene | 700-12-9 | ||
| Perylene | 198-55-0 | ||
| Phenacetin | 62-44-2 | ||
| cis-Permethrin | 61949-76-6 | ||
| trans-Permethrin | 61949-77-7 | ||
| Phenobarbital | 50-06-6 | ||
| Phenothiazene | 92-84-2 | ||
| 1,4-Phenylenediamine | 624-18-0 | ||
| 1-Phenylnaphthalene | 605-02-7 | ||
| 2-Phenylnaphthalene | 612-94-2 | ||
| Phorate | 298-02-2 | ||
| Phosalone | 2310-18-0 | ||
| Phosmet | 732-11-6 | ||
| Phosphamidon | 13171-21-6 | ||
| Phthalic anhydride | 85-44-9 | ||
| alpha-Picoline (2-Methylpyridine) | 109-06-8 | ||
| Piperonyl sulfoxide | 120-62-7 | ||
| Prometon | 1610-18-0 | ||
| Prometryn | 7287-19-6 | ||
| Pronamide | 23950-58-5 | ||
| Propachlor | 1918-16-7 | ||
| Propazine | 139-40-2 | ||
| Propylthiouracil | 51-52-5 | ||
| Pyridine | 110-86-1 | ||
| Resorcinol (1,3-Benzenediol) | 108-46-3 | ||
| Safrole | 94-59-7 | ||
| Simazine | 122-34-9 | ||
| Simetryn | 1014-70-6 | ||
| Squalene | 7683-64-9 | ||
| Stirofos | 22248-79-9 | ||
| Strychnine | 57-24-9 | ||
| Styrene 9 | 100-42-5 | ||
| Sulfallate | 95-06-7 | ||
| Tebuthiuron | 34014-18-1 | ||
| Terbacil | 5902-51-2 | ||
| Terbufos | 13071-79-9 | ||
| Terbutryn | 886-50-0 | ||
| alpha-Terpineol | 98-55-5 | ||
| 1,2,4,5-Tetrachlorobenzene | 95-94-3 | ||
| 2,2′,4,4′-Tetrachlorobiphenyl | 2437-79-8 | ||
| 2,3,7,8-Tetrachlorodibenzo-p-dioxin | 1746-01-6 | ||
| 2,3,4,6-Tetrachlorophenol | 58-90-2 | ||
| Tetrachlorvinphos | 22248-79-9 | ||
| Tetraethyl dithiopyrophosphate | 3689-24-5 | ||
| Tetraethyl pyrophosphate | 107-49-3 | ||
| Thianaphthene (2,3-Benzothiophene) | 95-15-8 | ||
| Thioacetamide | 62-55-5 | ||
| Thionazin | 297-97-2 | ||
| Thiophenol (Benzenethiol) | 108-98-5 | ||
| Thioxanthone | 492-22-8 | ||
| Toluene-1,3-diisocyanate | 26471-62-5 | ||
| Toluene-2,4-diisocyanate | 584-84-9 | ||
| o-Toluidine | 95-53-4 | ||
| Toxaphene 3 5 | 8001-35-2 | ||
| Triadimefon | 43121-43-3 | ||
| 1,2,3-Trichlorobenzene | 87-61-6 | ||
| 2,4,5-Trichlorobiphenyl | 15862-07-4 | ||
| 2,3,6-Trichlorophenol | 933-75-5 | ||
| 2,4,5-Trichlorophenol | 95-95-4 | ||
| Tricyclazole | 41814-78-2 | ||
| Trifluralin | 1582-09-8 | ||
| 1,2,3-Trimethoxybenzene | 634-36-6 | ||
| 2,4,5-Trimethylaniline | 137-17-7 | ||
| Trimethyl phosphate | 512-56-1 | ||
| Triphenylene | 217-59-4 | ||
| Tripropyleneglycolmethyl ether | 20324-33-8 | ||
| 1,3,5-Trinitrobenzene | 99-35-4 | ||
| Tris(2,3-dibromopropyl) phosphate | 126-72-7 | ||
| Tri-p-tolyl phosphate | 78-32-0 | ||
| O,O,O-Triethyl phosphorothioate | 126-68-1 | ||
| Trithiane | 291-29-4 | ||
| Vernolate | 1929-77-7 |
1 Compounds that have been demonstrated amenable to extraction and gas chromatography.
2 Determine each analyte in the fraction that gives the most accurate result.
3 Priority Pollutant (40 CFR part 423, appendix A).
4 See section 1.2.
5 These compounds are mixtures of various isomers.
6 Detected as azobenzene.
7 MDL values from the 1984 promulgated version of Method 625.
8 ML = Minimum Level - see Glossary for definition and derivation.
9 Styrene may be susceptible to losses during sampling, preservation, and/or extraction of full-volume (1 L) water samples. However, styrene is not regulated at 40 CFR part 136, and it is also listed as an analyte in EPA Method 624.1 and EPA Method 1625C, where such losses may be less than using Method 625.1.
Table 4 - Chromatographic Conditions and Characteristic m/z's for Base/Neutral Extractables
| Analyte | Retention
time (sec) 1 |
Characteristic m/z's | |||||
|---|---|---|---|---|---|---|---|
| Electron impact ionization | Chemical ionization | ||||||
| Primary | Second | Second | Methane | Methane | Methane | ||
| N-Nitrosodimethylamine | 385 | 42 | 74 | 44 | |||
| bis(2-Chloroethyl) ether | 704 | 93 | 63 | 95 | 63 | 107 | 109 |
| bis(2-Chloroisopropyl) ether | 799 | 45 | 77 | 79 | 77 | 135 | 137 |
| Hexachloroethane | 823 | 117 | 201 | 199 | 199 | 201 | 203 |
| N-Nitrosodi-n-propylamine | 830 | 130 | 42 | 101 | |||
| Nitrobenzene | 849 | 77 | 123 | 65 | 124 | 152 | 164 |
| Isophorone | 889 | 82 | 95 | 138 | 139 | 167 | 178 |
| bis(2-Chloroethoxy) methane | 939 | 93 | 95 | 123 | 65 | 107 | 137 |
| 1,2,4-Trichlorobenzene | 958 | 180 | 182 | 145 | 181 | 183 | 209 |
| Naphthalene | 967 | 128 | 129 | 127 | 129 | 157 | 169 |
| Hexachlorobutadiene | 1006 | 225 | 223 | 227 | 223 | 225 | 227 |
| Hexachlorocyclopentadiene | 1142 | 237 | 235 | 272 | 235 | 237 | 239 |
| 2-Chloronaphthalene | 1200 | 162 | 164 | 127 | 163 | 191 | 203 |
| Acenaphthylene | 1247 | 152 | 151 | 153 | 152 | 153 | 181 |
| Dimethyl phthalate | 1273 | 163 | 194 | 164 | 151 | 163 | 164 |
| 2,6-Dinitrotoluene | 1300 | 165 | 89 | 121 | 183 | 211 | 223 |
| Acenaphthene | 1304 | 154 | 153 | 152 | 154 | 155 | 183 |
| 2,4-Dinitrotoluene | 1364 | 165 | 63 | 182 | 183 | 211 | 223 |
| Fluorene | 1401 | 166 | 165 | 167 | 166 | 167 | 195 |
| 4-Chlorophenyl phenyl ether | 1409 | 204 | 206 | 141 | |||
| Diethyl phthalate | 1414 | 149 | 177 | 150 | 177 | 223 | 251 |
| N-Nitrosodiphenylamine | 1464 | 169 | 168 | 167 | 169 | 170 | 198 |
| 4-Bromophenyl phenyl ether | 1498 | 248 | 250 | 141 | 249 | 251 | 277 |
| alpha-BHC | 1514 | 183 | 181 | 109 | |||
| Hexachlorobenzene | 1522 | 284 | 142 | 249 | 284 | 286 | 288 |
| beta-BHC | 1544 | 183 | 181 | 109 | |||
| gamma-BHC | 1557 | 181 | 183 | 109 | |||
| Phenanthrene | 1583 | 178 | 179 | 176 | 178 | 179 | 207 |
| Anthracene | 1592 | 178 | 179 | 176 | 178 | 179 | 207 |
| delta-BHC | 1599 | 183 | 109 | 181 | |||
| Heptachlor | 1683 | 100 | 272 | 274 | |||
| Di-n-butyl phthalate | 1723 | 149 | 150 | 104 | 149 | 205 | 279 |
| Aldrin | 1753 | 66 | 263 | 220 | |||
| Fluoranthene | 1817 | 202 | 101 | 100 | 203 | 231 | 243 |
| Heptachlor epoxide | 1820 | 353 | 355 | 351 | |||
| gamma-Chlordane | 1834 | 373 | 375 | 377 | |||
| Pyrene | 1852 | 202 | 101 | 100 | 203 | 231 | 243 |
| Benzidine 2 | 1853 | 184 | 92 | 185 | 185 | 213 | 225 |
| alpha-Chlordane | 1854 | 373 | 375 | 377 | |||
| Endosulfan I | 1855 | 237 | 339 | 341 | |||
| 4,4′-DDE | 1892 | 246 | 248 | 176 | |||
| Dieldrin | 1907 | 79 | 263 | 279 | |||
| Endrin | 1935 | 81 | 263 | 82 | |||
| Endosulfan II | 2014 | 237 | 339 | 341 | |||
| 4,4′-DDD | 2019 | 235 | 237 | 165 | |||
| Endrin aldehyde | 2031 | 67 | 345 | 250 | |||
| Butyl benzyl phthalate | 2060 | 149 | 91 | 206 | 149 | 299 | 327 |
| Endosulfan sulfate | 2068 | 272 | 387 | 422 | |||
| 4,4′-DDT | 2073 | 235 | 237 | 165 | |||
| Chrysene | 2083 | 228 | 226 | 229 | 228 | 229 | 257 |
| 3,3′-Dichlorobenzidine | 2086 | 252 | 254 | 126 | |||
| Benzo(a)anthracene | 2090 | 228 | 229 | 226 | 228 | 229 | 257 |
| bis(2-Ethylhexyl) phthalate | 2124 | 149 | 167 | 279 | 149 | ||
| Di-n-octyl phthalate | 2240 | 149 | 43 | 57 | |||
| Benzo(b)fluoranthene | 2286 | 252 | 253 | 125 | 252 | 253 | 281 |
| Benzo(k)fluoranthene | 2293 | 252 | 253 | 125 | 252 | 253 | 281 |
| Benzo(a)pyrene | 2350 | 252 | 253 | 125 | 252 | 253 | 281 |
| Indeno(1,2,3-cd) pyrene | 2650 | 276 | 138 | 277 | 276 | 277 | 305 |
| Dibenz(a,h)anthracene | 2660 | 278 | 139 | 279 | 278 | 279 | 307 |
| Benzo(ghi)perylene | 2750 | 276 | 138 | 277 | 276 | 277 | 305 |
| Toxaphene | 159 | 231 | 233 | ||||
| PCB 1016 | 224 | 260 | 294 | ||||
| PCB 1221 | 190 | 224 | 260 | ||||
| PCB 1232 | 190 | 224 | 260 | ||||
| PCB 1242 | 224 | 260 | 294 | ||||
| PCB 1248 | 294 | 330 | 262 | ||||
| PCB 1254 | 294 | 330 | 362 | ||||
| PCB 1260 | 330 | 362 | 394 | ||||
1 Column: 30 m x 0.25 mm ID; 94% methyl, 5% phenyl, 1% vinyl bonded phase fused silica capillary.
Conditions: 5 min at 30 °C; 30-280 at 8 °C per min; isothermal at 280 °C until benzo(ghi)perylene elutes.
Gas velocity: 30 cm/sec at 30 °C (at constant pressure).
2 See section 1.2; included for tailing factor testing.
Table 5 - Chromatographic Conditions and Characteristic m/z's for Acid Extractables
| Analyte | Retention
Time (sec) 1 |
Characteristic m/z's | |||||
|---|---|---|---|---|---|---|---|
| Electron impact ionization | Chemical ionization | ||||||
| Prime | Second | Second | Methane | Methane | Methane | ||
| 2-Chlorophenol | 705 | 128 | 64 | 130 | 129 | 131 | 157 |
| Phenol | 700 | 94 | 65 | 66 | 95 | 123 | 135 |
| 2-Nitrophenol | 900 | 139 | 65 | 109 | 140 | 168 | 122 |
| 2,4-Dimethylphenol | 924 | 122 | 107 | 121 | 123 | 151 | 163 |
| 2,4-Dichlorophenol | 947 | 162 | 164 | 98 | 163 | 165 | 167 |
| 4-Chloro-3-methylphenol | 1091 | 142 | 107 | 144 | 143 | 171 | 183 |
| 2,4,6-Trichlorophenol | 1165 | 196 | 198 | 200 | 197 | 199 | 201 |
| 2,4-Dinitrophenol | 1325 | 184 | 63 | 154 | 185 | 213 | 225 |
| 4-Nitrophenol | 1354 | 65 | 139 | 109 | 140 | 168 | 122 |
| 2-Methyl-4,6-dinitrophenol | 1435 | 198 | 182 | 77 | 199 | 227 | 239 |
| Pentachlorophenol | 1561 | 266 | 264 | 268 | 267 | 265 | 269 |
Column: 30 m x 0.25 mm ID; 94% methyl, 5% phenyl, 1% vinyl bonded phase fused silica capillary.
Conditions: 5 min at 30 °C; 30-250 at 8 °C per min; isothermal at 280 °C until pentachlorophenol elutes.
Gas velocity: 30 cm/sec at 30 °C (at constant pressure).
Table 6 - QC Acceptance Criteria - Method 625 1
| Analyte | Range for Q (%) 2 |
Limit for s (%) 3 |
Range for X (%) 3 |
Range for P1, P2(%) 3 |
Limit for RPD (%) |
|---|---|---|---|---|---|
| Acenaphthene | 70-130 | 29 | 60-132 | 47-145 | 48 |
| Acenaphthylene | 60-130 | 45 | 54-126 | 33-145 | 74 |
| Aldrin | 7-152 | 39 | 7-152 | D-166 | 81 |
| Anthracene | 58-130 | 40 | 43-120 | 27-133 | 66 |
| Benzo(a)anthracene | 42-133 | 32 | 42-133 | 33-143 | 53 |
| Benzo(b)fluoranthene | 42-140 | 43 | 42-140 | 24-159 | 71 |
| Benzo(k)fluoranthene | 25-146 | 38 | 25-146 | 11-162 | 63 |
| Benzo(a)pyrene | 32-148 | 43 | 32-148 | 17-163 | 72 |
| Benzo(ghi)perylene | 13-195 | 61 | D-195 | D-219 | 97 |
| Benzyl butyl phthalate | 43-140 | 36 | D-140 | D-152 | 60 |
| beta-BHC | 42-131 | 37 | 42-131 | 24-149 | 61 |
| delta-BHC | D-130 | 77 | D-120 | D-120 | 129 |
| bis(2-Chloroethyl)ether | 52-130 | 65 | 43-126 | 12-158 | 108 |
| bis(2-Chloroethoxy)methane | 52-164 | 32 | 49-165 | 33-184 | 54 |
| bis(2-Chloroisopropyl) ether | 63-139 | 46 | 63-139 | 36-166 | 76 |
| bis(2-Ethylhexyl) phthalate | 43-137 | 50 | 29-137 | 8-158 | 82 |
| 4-Bromophenyl phenyl ether | 70-130 | 26 | 65-120 | 53-127 | 43 |
| 2-Chloronaphthalene | 70-130 | 15 | 65-120 | 60-120 | 24 |
| 4-Chlorophenyl phenyl ether | 57-145 | 36 | 38-145 | 25-158 | 61 |
| Chrysene | 44-140 | 53 | 44-140 | 17-168 | 87 |
| 4,4′-DDD | D-135 | 56 | D-135 | D-145 | 93 |
| 4,4′-DDE | 19-130 | 46 | 19-120 | 4-136 | 77 |
| 4,4′-DDT | D-171 | 81 | D-171 | D-203 | 135 |
| Dibenz(a,h)anthracene | 13-200 | 75 | D-200 | D-227 | 126 |
| Di-n-butyl phthalate | 52-130 | 28 | 8-120 | 1-120 | 47 |
| 3,3′-Dichlorobenzidine | 18-213 | 65 | 8-213 | D-262 | 108 |
| Dieldrin | 70-130 | 38 | 44-119 | 29-136 | 62 |
| Diethyl phthalate | 47-130 | 60 | D-120 | D-120 | 100 |
| Dimethyl phthalate | 50-130 | 110 | D-120 | D-120 | 183 |
| 2,4-Dinitrotoluene | 53-130 | 25 | 48-127 | 39-139 | 42 |
| 2,6-Dinitrotoluene | 68-137 | 29 | 68-137 | 50-158 | 48 |
| Di-n-octyl phthalate | 21-132 | 42 | 19-132 | 4-146 | 69 |
| Endosulfan sulfate | D-130 | 42 | D-120 | D-120 | 70 |
| Endrin aldehyde | D-189 | 45 | D-189 | D-209 | 75 |
| Fluoranthene | 47-130 | 40 | 43-121 | 26-137 | 66 |
| Fluorene | 70-130 | 23 | 70-120 | 59-121 | 38 |
| Heptachlor | D-172 | 44 | D-172 | D-192 | 74 |
| Heptachlor epoxide | 70-130 | 61 | 71-120 | 26-155 | 101 |
| Hexachlorobenzene | 38-142 | 33 | 8-142 | D-152 | 55 |
| Hexachlorobutadiene | 68-130 | 38 | 38-120 | 24-120 | 62 |
| Hexachloroethane | 55-130 | 32 | 55-120 | 40-120 | 52 |
| Indeno(1,2,3-cd)pyrene | 13-151 | 60 | D-151 | D-171 | 99 |
| Isophorone | 52-180 | 56 | 47-180 | 21-196 | 93 |
| Naphthalene | 70-130 | 39 | 36-120 | 21-133 | 65 |
| Nitrobenzene | 54-158 | 37 | 54-158 | 35-180 | 62 |
| N-Nitrosodi-n-propylamine | 59-170 | 52 | 14-198 | D-230 | 87 |
| PCB-1260 | 19-130 | 77 | 19-130 | D-164 | 128 |
| Phenanthrene | 67-130 | 24 | 65-120 | 54-120 | 39 |
| Pyrene | 70-130 | 30 | 70-120 | 52-120 | 49 |
| 1,2,4-Trichlorobenzene | 61-130 | 30 | 57-130 | 44-142 | 50 |
| 4-Chloro-3-methylphenol | 68-130 | 44 | 41-128 | 22-147 | 73 |
| 2-Chlorophenol | 55-130 | 37 | 36-120 | 23-134 | 61 |
| 2,4-Dichlorophenol | 64-130 | 30 | 53-122 | 39-135 | 50 |
| 2,4-Dimethylphenol | 58-130 | 35 | 42-120 | 32-120 | 58 |
| 2,4-Dinitrophenol | 39-173 | 79 | D-173 | D-191 | 132 |
| 2-Methyl-4,6-dinitrophenol | 56-130 | 122 | 53-130 | D-181 | 203 |
| 2-Nitrophenol | 61-163 | 33 | 45-167 | 29-182 | 55 |
| 4-Nitrophenol | 35-130 | 79 | 13-129 | D-132 | 131 |
| Pentachlorophenol | 42-152 | 52 | 38-152 | 14-176 | 86 |
| Phenol | 48-130 | 39 | 17-120 | 5-120 | 64 |
| 2,4,6-Trichlorophenol | 69-130 | 35 | 52-129 | 37-144 | 58 |
1 Acceptance criteria are based upon method performance data in Table 7 and from EPA Method 1625. Where necessary, limits for recovery have been broadened to assure applicability to concentrations below those used to develop Table 7.
2 Test concentration = 100 µg/mL.
3 Test concentration = 100 µg/L.
Q = Calibration verification (sections 7.3.1 and 13.4).
s = Standard deviation for four recovery measurements in the DOC test (section 8.2.4).
X = Average recovery for four recovery measurements in the DOC test (section 8.2.4).
P1, P2 = MS/MSD recovery (section 8.3.2, section 8.4.2).
RPD = MS/MSD relative percent difference (RPD; section 8.3.3).
D = Detected; result must be greater than zero.
Table 7 - Precision and Recovery as Functions of Concentration - Method 625 1
| Analyte | Recovery, X′ (µg/L) |
Single analyst precision, sr′ (µg/L) |
Overall precision, S′ (µg/L) |
|---|---|---|---|
| Acenaphthene | 0.96C + 0.19 | 0.15 X−0.12 | 0.21 X−0.67 |
| Acenaphthylene | 0.89C + 0.74 | 0.24 X−1.06 | 0.26 X−0.54 |
| Aldrin | 0.78C + 1.66 | 0.27 X−1.28 | 0.43 X + 1.13 |
| Anthracene | 0.80C + 0.68 | 0.21 X−0.32 | 0.27 X−0.64 |
| Benzo(a)anthracene | 0.88C−0.60 | 0.15 X + 0.93 | 0.26 X−0.28 |
| Benzo(b)fluoranthene | 0.93C−1.80 | 0.22 X + 0.43 | 0.29 X + 0.96 |
| Benzo(k)fluoranthene | 0.87C−1.56 | 0.19 X + 1.03 | 0.35 X + 0.40 |
| Benzo(a)pyrene | 0.90C−0.13 | 0.22 X + 0.48 | 0.32 X + 1.35 |
| Benzo(ghi)perylene | 0.98C−0.86 | 0.29 X + 2.40 | 0.51 X−0.44 |
| Benzyl butyl phthalate | 0.66C−1.68 | 0.18 X + 0.94 | 0.53 X + 0.92 |
| beta-BHC | 0.87C−0.94 | 0.20 X−0.58 | 0.30 X−1.94 |
| delta-BHC | 0.29C−1.09 | 0.34 X + 0.86 | 0.93 X−0.17 |
| bis(2-Chloroethyl) ether | 0.86C−1.54 | 0.35 X−0.99 | 0.35 X + 0.10 |
| bis(2-Chloroethoxy) methane | 1.12C−5.04 | 0.16 X + 1.34 | 0.26 X + 2.01 |
| bis(2-Chloroisopropyl) ether | 1.03C−2.31 | 0.24 X + 0.28 | 0.25 X + 1.04 |
| bis(2-Ethylhexyl) phthalate | 0.84C−1.18 | 0.26 X + 0.73 | 0.36 X + 0.67 |
| 4-Bromophenyl phenyl ether | 0.91C−1.34 | 0.13 X + 0.66 | 0.16 X + 0.66 |
| 2-Chloronaphthalene | 0.89C + 0.01 | 0.07 X + 0.52 | 0.13 X + 0.34 |
| 4-Chlorophenyl phenyl ether | 0.91C + 0.53 | 0.20 X−0.94 | 0.30 X−0.46 |
| Chrysene | 0.93C−1.00 | 0.28 X + 0.13 | 0.33 X−0.09 |
| 4,4′-DDD | 0.56C−0.40 | 0.29 X−0.32 | 0.66 X−0.96 |
| 4,4′-DDE | 0.70C−0.54 | 0.26 X−1.17 | 0.39 X−1.04 |
| 4,4′-DDT | 0.79C−3.28 | 0.42 X + 0.19 | 0.65 X−0.58 |
| Dibenz(a,h)anthracene | 0.88C + 4.72 | 0.30 X + 8.51 | 0.59 X + 0.25 |
| Di-n-butyl phthalate | 0.59C + 0.71 | 0.13 X + 1.16 | 0.39 X + 0.60 |
| 3,3'-Dichlorobenzidine | 1.23C−12.65 | 0.28 X + 7.33 | 0.47 X + 3.45 |
| Dieldrin | 0.82C−0.16 | 0.20 X−0.16 | 0.26 X−0.07 |
| Diethyl phthalate | 0.43C + 1.00 | 0.28 X + 1.44 | 0.52 X + 0.22 |
| Dimethyl phthalate | 0.20C + 1.03 | 0.54 X + 0.19 | 1.05 X−0.92 |
| 2,4-Dinitrotoluene | 0.92C−4.81 | 0.12 X + 1.06 | 0.21 X + 1.50 |
| 2,6-Dinitrotoluene | 1.06C−3.60 | 0.14 X + 1.26 | 0.19 X + 0.35 |
| Di-n-octyl phthalate | 0.76C−0.79 | 0.21 X + 1.19 | 0.37 X + 1.19 |
| Endosulfan sulfate | 0.39C + 0.41 | 0.12 X + 2.47 | 0.63 X−1.03 |
| Endrin aldehyde | 0.76C−3.86 | 0.18 X + 3.91 | 0.73 X−0.62 |
| Fluoranthene | 0.81C + 1.10 | 0.22 X + 0.73 | 0.28 X−0.60 |
| Fluorene | 0.90C−0.00 | 0.12 X + 0.26 | 0.13 X + 0.61 |
| Heptachlor | 0.87C−2.97 | 0.24 X−0.56 | 0.50 X−0.23 |
| Heptachlor epoxide | 0.92C−1.87 | 0.33 X−0.46 | 0.28 X + 0.64 |
| Hexachlorobenzene | 0.74C + 0.66 | 0.18 X−0.10 | 0.43 X−0.52 |
| Hexachlorobutadiene | 0.71C−1.01 | 0.19 X + 0.92 | 0.26 X + 0.49 |
| Hexachloroethane | 0.73C−0.83 | 0.17 X + 0.67 | 0.17 X + 0.80 |
| Indeno(1,2,3-cd)pyrene | 0.78C−3.10 | 0.29 X + 1.46 | 0.50 X + 0.44 |
| Isophorone | 1.12C + 1.41 | 0.27 X + 0.77 | 0.33 X + 0.26 |
| Naphthalene | 0.76C + 1.58 | 0.21 X−0.41 | 0.30 X−0.68 |
| Nitrobenzene | 1.09C−3.05 | 0.19 X + 0.92 | 0.27 X + 0.21 |
| N-Nitrosodi-n-propylamine | 1.12C−6.22 | 0.27 X + 0.68 | 0.44 X + 0.47 |
| PCB-1260 | 0.81C−10.86 | 0.35 X + 3.61 | 0.43 X + 1.82 |
| Phenanthrene | 0.87C−0.06 | 0.12 X + 0.57 | 0.15 X + 0.25 |
| Pyrene | 0.84C−0.16 | 0.16 X + 0.06 | 0.15 X + 0.31 |
| 1,2,4-Trichlorobenzene | 0.94C−0.79 | 0.15 X + 0.85 | 0.21 X + 0.39 |
| 4-Chloro-3-methylphenol | 0.84C + 0.35 | 0.23 X + 0.75 | 0.29 X + 1.31 |
| 2-Chlorophenol | 0.78C + 0.29 | 0.18 X + 1.46 | 0.28 X + 0.97 |
| 2,4-Dichlorophenol | 0.87C + 0.13 | 0.15 X + 1.25 | 0.21 X + 1.28 |
| 2,4-Dimethylphenol | 0.71C + 4.41 | 0.16 X + 1.21 | 0.22 X + 1.31 |
| 2,4-Dinitrophenol | 0.81C−18.04 | 0.38 X + 2.36 | 0.42 X + 26.29 |
| 2-Methyl-4,6-Dinitrophenol | 1.04C−28.04 | 0.05 X + 42.29 | 0.26 X + 23.10 |
| 2-Nitrophenol | 1.07C−1.15 | 0.16 X + 1.94 | 0.27 X + 2.60 |
| 4-Nitrophenol | 0.61C−1.22 | 0.38 X + 2.57 | 0.44 X + 3.24 |
| Pentachlorophenol | 0.93C + 1.99 | 0.24 X + 3.03 | 0.30 X + 4.33 |
| Phenol | 0.43C + 1.26 | 0.26 X + 0.73 | 0.35 X + 0.58 |
| 2,4,6-Trichlorophenol | 0.91C−0.18 | 0.16 X + 2.22 | 0.22 X + 1.81 |
1 Regressions based on data from Reference 2.
X′ = Expected recovery for one or more measurements of a sample containing a concentration of C, in µg/L.
sr′ = Expected single analyst standard deviation of measurements at an average concentration found of X, in µg/L.
S′ = Expected interlaboratory standard deviation of measurements at an average concentration found of X, in µg/L.
C = True value for the concentration, in µg/L.
X = Average recovery found for measurements of samples containing a concentration of C, in µg/L.
Table 8 - Suggested Internal and Surrogate Standards
| Base/neutral fraction | Range for surrogate recovery (%) 1 | |
|---|---|---|
| Calibration verification | Recovery from samples | |
| Acenaphthalene-d8 | 66-152 | 33-168 |
| Acenaphthene-d10 | 71-141 | 30-180 |
| Aniline-d5 | ||
| Anthracene-d10 | 58-171 | 23-142 |
| Benzo(a)anthracene-d12 | 28-357 | 22-329 |
| Benzo(a)pyrene-d12 | 32-194 | 32-194 |
| 4-Chloroaniline-d4 | 1-145 | 1-145 |
| bis(2-Chloroethyl) ether-d8 | 52-194 | 25-222 |
| Chrysene-d12 | 23-290 | 23-290 |
| Decafluorobiphenyl | ||
| 4,4′-Dibromobiphenyl | ||
| 4,4′-Dibromooctafluorobiphenyl | ||
| 1,4-Dichlorobenzene-d4 | 65-153 | 11-245 |
| 2,2′-Difluorobiphenyl | ||
| Dimethyl phthalate-d6 | 47-211 | 1-500 |
| Fluoranthene-d10 | 47-215 | 30-187 |
| Fluorene-d10 | 61-164 | 38-172 |
| 4-Fluoroaniline | ||
| 1-Fluoronaphthalene | ||
| 2-Fluoronaphthalene | ||
| 2-Methylnaphthalene-d10 | 50-150 | 50-150 |
| Naphthalene-d8 | 71-141 | 22-192 |
| Nitrobenzene-d5 | 46-219 | 15-314 |
| 2,3,4,5,6-Pentafluorobiphenyl | ||
| Perylene-d12 | ||
| Phenanthrene-d10 | 67-149 | 34-168 |
| Pyrene-d10 | 48-210 | 28-196 |
| Pyridine-d5 | ||
| Acid fraction | ||
| 2-Chlorophenol-d4 | 55-180 | 33-180 |
| 2,4-Dichlorophenol-d3 | 64-157 | 34-182 |
| 4,6-Dinitro-2-methylphenol-d2 | 56-177 | 22-307 |
| 2-Fluorophenol | ||
| 4-Methylphenol-d8 | 25-111 | 25-111 |
| 2-Nitrophenol-d4 | 61-163 | 37-163 |
| 4-Nitrophenol-d4 | 35-287 | 6-500 |
| Pentafluorophenol | ||
| 2-Perfluoromethylphenol | ||
| Phenol-d5 | 48-208 | 8-424 |
1 Recovery from samples is the wider of the criteria in the CLP SOW for organics or in Method 1625.
Table 9A - DFTPP Key m/z's and Abundance Criteria for Quadrupole Instruments 1
| m/z | Abundance criteria |
|---|---|
| 51 | 30-60 percent of m/z 198. |
| 68 | Less than 2 percent of m/z 69. |
| 70 | Less than 2 percent of m/z 69. |
| 127 | 40-60 percent of base peak m/z 198. |
| 197 | Less than 1 percent of m/z 198. |
| 198 | Base peak, 100 percent relative abundance. |
| 199 | 5-9 percent of m/z 198. |
| 275 | 10-30 percent of m/z 198. |
| 365 | Greater than 1 percent of m/z 198. |
| 441 | Present but less than m/z 443. |
| 442 | 40-100 percent of m/z 198. |
| 443 | 17-23 percent of m/z 442. |
1 Criteria in these tables are for quadrupole and time-of-flight instruments. Alternative tuning criteria from other published EPA reference methods may be used provided method performance is not adversely affected. Alternative tuning criteria specified by an instrument manufacturer may also be used for another type of mass spectrometer, provided method performance is not adversely affected.
Table 9B - DFTPP Key m/z's and Abundance Criteria for Time-of-flight Instruments 1
| m/z | Abundance criteria |
|---|---|
| 51 | 10-85 percent of the base peak. |
| 68 | Less than 2 percent of m/z 69. |
| 70 | Less than 2 percent of m/z 69. |
| 127 | 10-80 percent of the base peak. |
| 197 | Less than 2 percent of Mass 198. |
| 198 | Base peak, or greater than 50% of m/z 442. |
| 199 | 5-9 percent of m/z 198. |
| 275 | 10-60 percent of the base peak. |
| 365 | Greater than 0.5 percent of m/z 198. |
| 441 | Less than 150 percent of m/z 443. |
| 442 | Base peak or greater than 30 percent of m/z 198. |
| 443 | 15-24 percent of m/z 442. |
1 Criteria in these tables are for quadrupole and time-of-flight instruments. Alternative tuning criteria from other published EPA reference methods may be used provided method performance is not adversely affected. Alternative tuning criteria specified by an instrument manufacturer may also be used for another type of mass spectrometer, or for an alternative carrier gas, provided method performance is not adversely affected.
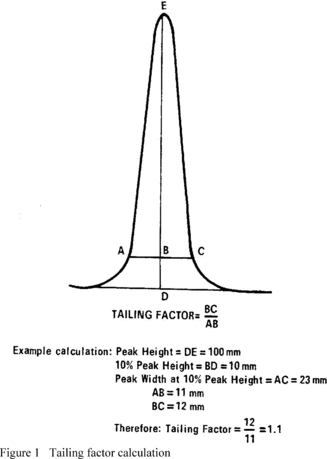
 22. Glossary
22. Glossary
These definitions and purposes are specific to this method but have been conformed to common usage to the extent possible.
22.1 Units of weight and measure and their abbreviations.
22.1.1 Symbols.
°C degrees Celsius µg microgram µL microliter < less than > greater than ≤ less than or equal to % percent22.1.2 Abbreviations (in alphabetical order).
cm centimeter g gram h hour ID inside diameter in. inch L liter m mass or meter mg milligram min minute mL milliliter mm millimeter ms millisecond m/z mass-to-charge ratio N normal; gram molecular weight of solute divided by hydrogen equivalent of solute, per liter of solution ng nanogram pg picogram ppb part-per-billion ppm part-per-million ppt part-per-trillion psig pounds-per-square inch gauge22.2 Definitions and acronyms (in alphabetical order).
Analyte - A compound or mixture of compounds (e.g., PCBs) tested for by this method. The analytes are listed in Tables 1-3.
Batch - See Extraction.
Blank - An aliquot of reagent water that is treated exactly as a sample including exposure to all glassware, equipment, solvents, reagents, internal standards, and surrogates that are used with samples. The blank is used to determine if analytes or interferences are present in the laboratory environment, the reagents, or the apparatus.
Calibration - The process of determining the relationship between the output or response of a measuring instrument and the value of an input standard. Historically, EPA has referred to a multi-point calibration as the “initial calibration,” to differentiate it from a single-point calibration verification.
Calibration standard - A solution prepared from stock solutions and/or a secondary standards and containing the analytes of interest, surrogates, and internal standards. The calibration standard is used to calibrate the response of the GC/MS instrument against analyte concentration.
Calibration verification standard - The mid-point calibration standard used to verify calibration. See sections 7.3 and 13.4.
Descriptor - In SIM, the beginning and ending retention times for the RT window, the m/z's sampled in the RT window, and the dwell time at each m/z.
Extracted ion current profile (EICP) - The line described by the signal at a given m/z.
Extraction Batch - A set of up to 20 field samples (not including QC samples) started through the extraction process on a given 24-hour shift (section 3.1). Each extraction batch must be accompanied by a blank (section 8.5), a laboratory control sample (LCS, section 8.4), and a matrix spike and duplicate (MS/MSD; Section 8.3), resulting in a minimum of five analyses (1 sample, 1 blank, 1 LCS, 1 MS, and 1 MSD) and a maximum of 24 analyses (20 field samples, 1 blank, 1 LCS, 1 MS, and 1 MSD) for the batch. If greater than 20 samples are to be extracted in a 24-hour shift, the samples must be separated into extraction batches of 20 or fewer samples.
Field Duplicates - Two samples collected at the same time and placed under identical conditions, and treated identically throughout field and laboratory procedures. Results of analyses of the field duplicates provide an estimate of the precision associated with sample collection, preservation, and storage, as well as with laboratory procedures.
Field blank - An aliquot of reagent water or other reference matrix that is placed in a sample container in the field, and treated as a sample in all respects, including exposure to sampling site conditions, storage, preservation, and all analytical procedures. The purpose of the field blank is to determine if the field or sample transporting procedures and environments have contaminated the sample.
GC - Gas chromatograph or gas chromatography.
Internal standard - A compound added to an extract or standard solution in a known amount and used as a reference for quantitation of the analytes of interest and surrogates. In this method the internal standards are stable isotopically labeled analogs of selected method analytes (Table 8). Also see Internal standard quantitation.
Internal standard quantitation - A means of determining the concentration of an analyte of interest (Tables 1-3) by reference to a compound not expected to be found in a sample.
DOC - Initial demonstration of capability (section 8.2); four aliquots of reagent water spiked with the analytes of interest and analyzed to establish the ability of the laboratory to generate acceptable precision and recovery. A DOC is performed prior to the first time this method is used and any time the method or instrumentation is modified.
Laboratory Control Sample (LCS; laboratory fortified blank; section 8.4) - An aliquot of reagent water spiked with known quantities of the analytes of interest and surrogates. The LCS is analyzed exactly like a sample. Its purpose is to assure that the results produced by the laboratory remain within the limits specified in this method for precision and recovery.
Laboratory fortified sample matrix - See Matrix spike.
Laboratory reagent blank - A blank run on laboratory reagents; e.g., methylene chloride (section 11.1.5).
Matrix spike (MS) and matrix spike duplicate (MSD) (laboratory fortified sample matrix and duplicate) - Two aliquots of an environmental sample to which known quantities of the analytes of interest and surrogates are added in the laboratory. The MS/MSD are prepared and analyzed exactly like a field sample. Their purpose is to quantify any additional bias and imprecision caused by the sample matrix. The background concentrations of the analytes in the sample matrix must be determined in a separate aliquot and the measured values in the MS/MSD corrected for background concentrations.
May - This action, activity, or procedural step is neither required nor prohibited.
May not - This action, activity, or procedural step is prohibited.
Method blank - See blank.
Method detection limit (MDL) - A detection limit determined by the procedure at 40 CFR part 136, appendix B. The MDLs determined by EPA in the original version of the method are listed in Tables 1, 2 and 3. As noted in section 1.5, use the MDLs in Tables 1, 2, and 3 in conjunction with current MDL data from the laboratory actually analyzing samples to assess the sensitivity of this procedure relative to project objectives and regulatory requirements (where applicable).
Minimum level (ML) - The term “minimum level” refers to either the sample concentration equivalent to the lowest calibration point in a method or a multiple of the method detection limit (MDL), whichever is higher. Minimum levels may be obtained in several ways: They may be published in a method; they may be based on the lowest acceptable calibration point used by a laboratory; or they may be calculated by multiplying the MDL in a method, or the MDL determined by a laboratory, by a factor of 3. For the purposes of NPDES compliance monitoring, EPA considers the following terms to be synonymous: “quantitation limit,” “reporting limit,” and “minimum level.”
MS - Mass spectrometer or mass spectrometry, or matrix spike (a QC sample type).
MSD - Matrix spike duplicate (a QC sample type).
Must - This action, activity, or procedural step is required.
m/z - The ratio of the mass of an ion (m) detected in the mass spectrometer to the charge (z) of that ion.
Preparation blank - See blank.
Quality control check sample (QCS) - See Laboratory Control Sample.
Reagent water - Water demonstrated to be free from the analytes of interest and potentially interfering substances at the MDLs for the analytes in this method.
Regulatory compliance limit (or regulatory concentration limit) - A limit on the concentration or amount of a pollutant or contaminant specified in a nationwide standard, in a permit, or otherwise established by a regulatory/control authority.
Relative retention time (RRT) - The ratio of the retention time of an analyte to the retention time of its associated internal standard. RRT compensates for small changes in the GC temperature program that can affect the absolute retention times of the analyte and internal standard. RRT is a unitless quantity.
Relative standard deviation (RSD) - The standard deviation times 100 divided by the mean. Also termed “coefficient of variation.”
RF - Response factor. See section 7.2.2.
RSD - See relative standard deviation.
Safety Data Sheet (SDS) - Written information on a chemical's toxicity, health hazards, physical properties, fire, and reactivity, including storage, spill, and handling precautions that meet the requirements of OSHA, 29 CFR 1910.1200(g) and appendix D to § 1910.1200. United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS), third revised edition, United Nations, 2009.
Selected Ion Monitoring (SIM) - An MS technique in which a few m/z's are monitored. When used with gas chromatography, the m/z's monitored are usually changed periodically throughout the chromatographic run, to correlate with the characteristic m/z's of the analytes, surrogates, and internal standards as they elute from the chromatographic column. The technique is often used to increase sensitivity and minimize interferences.
Signal-to-noise ratio (S/N) - The height of the signal as measured from the mean (average) of the noise to the peak maximum divided by the width of the noise.
Should - This action, activity, or procedural step is suggested but not required.
SPE - Solid-phase extraction; an extraction technique in which an analyte is extracted from an aqueous solution by passage over or through a material capable of reversibly adsorbing the analyte. Also termed liquid-solid extraction.
Stock solution - A solution containing an analyte that is prepared using a reference material traceable to EPA, the National Institute of Science and Technology (NIST), or a source that will attest to the purity, authenticity, and concentration of the standard.
Surrogate - A compound unlikely to be found in a sample, and which is spiked into sample in a known amount before extraction or other processing, and is quantitated with the same procedures used to quantify other sample components. The purpose of the surrogate is to monitor method performance with each sample.
Method 1613, Revision B Tetra- Through Octa-Chlorinated Dioxins and Furans by Isotope Dilution HRGC/HRMS 1.0 Scope and Application1.1 This method is for determination of tetra- through octa-chlorinated dibenzo-p-dioxins (CDDs) and dibenzofurans (CDFs) in water, soil, sediment, sludge, tissue, and other sample matrices by high resolution gas chromatography/high resolution mass spectrometry (HRGC/HRMS). The method is for use in EPA's data gathering and monitoring programs associated with the Clean Water Act, the Resource Conservation and Recovery Act, the Comprehensive Environmental Response, Compensation and Liability Act, and the Safe Drinking Water Act. The method is based on a compilation of EPA, industry, commercial laboratory, and academic methods (References 1-6).
1.2 The seventeen 2,3,7,8-substituted CDDs/CDFs listed in Table 1 may be determined by this method. Specifications are also provided for separate determination of 2,3,7,8-tetrachloro-dibenzo-p-dioxin (2,3,7,8-TCDD) and 2,3,7,8-tetrachloro-dibenzofuran (2,3,7,8-TCDF).
1.3 The detection limits and quantitation levels in this method are usually dependent on the level of interferences rather than instrumental limitations. The minimum levels (MLs) in Table 2 are the levels at which the CDDs/CDFs can be determined with no interferences present. The Method Detection Limit (MDL) for 2,3,7,8-TCDD has been determined as 4.4 pg/L (parts-per-quadrillion) using this method.
1.4 The GC/MS portions of this method are for use only by analysts experienced with HRGC/HRMS or under the close supervision of such qualified persons. Each laboratory that uses this method must demonstrate the ability to generate acceptable results using the procedure in Section 9.2.
1.5 This method is “performance-based”. The analyst is permitted to modify the method to overcome interferences or lower the cost of measurements, provided that all performance criteria in this method are met. The requirements for establishing method equivalency are given in Section 9.1.2.
1.6 Any modification of this method, beyond those expressly permitted, shall be considered a major modification subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
2.0 Summary of MethodFlow charts that summarize procedures for sample preparation, extraction, and analysis are given in Figure 1 for aqueous and solid samples, Figure 2 for multi-phase samples, and Figure 3 for tissue samples.
2.1 Extraction.
2.1.1 Aqueous samples (samples containing less than 1% solids) - Stable isotopically labeled analogs of 15 of the 2,3,7,8-substituted CDDs/CDFs are spiked into a 1 L sample, and the sample is extracted by one of three procedures:
2.1.1.1 Samples containing no visible particles are extracted with methylene chloride in a separatory funnel or by the solid-phase extraction technique summarized in Section 2.1.1.3. The extract is concentrated for cleanup.
2.1.1.2 Samples containing visible particles are vacuum filtered through a glass-fiber filter. The filter is extracted in a Soxhlet/Dean-Stark (SDS) extractor (Reference 7), and the filtrate is extracted with methylene chloride in a separatory funnel. The methylene chloride extract is concentrated and combined with the SDS extract prior to cleanup.
2.1.1.3 The sample is vacuum filtered through a glass-fiber filter on top of a solid-phase extraction (SPE) disk. The filter and disk are extracted in an SDS extractor, and the extract is concentrated for cleanup.
2.1.2 Solid, semi-solid, and multi-phase samples (but not tissue) - The labeled compounds are spiked into a sample containing 10 g (dry weight) of solids. Samples containing multiple phases are pressure filtered and any aqueous liquid is discarded. Coarse solids are ground or homogenized. Any non-aqueous liquid from multi-phase samples is combined with the solids and extracted in an SDS extractor. The extract is concentrated for cleanup.
2.1.3 Fish and other tissue - The sample is extracted by one of two procedures:
2.1.3.1 Soxhlet or SDS extraction - A 20 g aliquot of sample is homogenized, and a 10 g aliquot is spiked with the labeled compounds. The sample is mixed with sodium sulfate, allowed to dry for 12-24 hours, and extracted for 18-24 hours using methylene chloride:hexane (1:1) in a Soxhlet extractor. The extract is evaporated to dryness, and the lipid content is determined.
2.1.3.2 HCl digestion - A 20 g aliquot is homogenized, and a 10 g aliquot is placed in a bottle and spiked with the labeled compounds. After equilibration, 200 mL of hydrochloric acid and 200 mL of methylene chloride:hexane (1:1) are added, and the bottle is agitated for 12-24 hours. The extract is evaporated to dryness, and the lipid content is determined.
2.2 After extraction, 37Cl4-labeled 2,3,7,8-TCDD is added to each extract to measure the efficiency of the cleanup process. Sample cleanups may include back-extraction with acid and/or base, and gel permeation, alumina, silica gel, Florisil and activated carbon chromatography. High-performance liquid chromatography (HPLC) can be used for further isolation of the 2,3,7,8-isomers or other specific isomers or congeners. Prior to the cleanup procedures cited above, tissue extracts are cleaned up using an anthropogenic isolation column, a batch silica gel adsorption, or sulfuric acid and base back-extraction, depending on the tissue extraction procedure used.
2.3 After cleanup, the extract is concentrated to near dryness. Immediately prior to injection, internal standards are added to each extract, and an aliquot of the extract is injected into the gas chromatograph. The analytes are separated by the GC and detected by a high-resolution (≥10,000) mass spectrometer. Two exact m/z's are monitored for each analyte.
2.4 An individual CDD/CDF is identified by comparing the GC retention time and ion-abundance ratio of two exact m/z's with the corresponding retention time of an authentic standard and the theoretical or acquired ion-abundance ratio of the two exact m/z's. The non-2,3,7,8 substituted isomers and congeners are identified when retention times and ion-abundance ratios agree within predefined limits. Isomer specificity for 2,3,7,8-TCDD and 2,3,7,8-TCDF is achieved using GC columns that resolve these isomers from the other tetra-isomers.
2.5 Quantitative analysis is performed using selected ion current profile (SICP) areas, in one of three ways:
2.5.1 For the 15 2,3,7,8-substituted CDDs/CDFs with labeled analogs (see Table 1), the GC/MS system is calibrated, and the concentration of each compound is determined using the isotope dilution technique.
2.5.2 For 1,2,3,7,8,9-HxCDD, OCDF, and the labeled compounds, the GC/MS system is calibrated and the concentration of each compound is determined using the internal standard technique.
2.5.3 For non-2,3,7,8-substituted isomers and for all isomers at a given level of chlorination (i.e., total TCDD), concentrations are determined using response factors from calibration of the CDDs/CDFs at the same level of chlorination.
2.6 The quality of the analysis is assured through reproducible calibration and testing of the extraction, cleanup, and GC/MS systems.
3.0 DefinitionsDefinitions are given in the glossary at the end of this method.
4.0 Contamination and Interferences4.1 Solvents, reagents, glassware, and other sample processing hardware may yield artifacts and/or elevated baselines causing misinterpretation of chromatograms (References 8-9). Specific selection of reagents and purification of solvents by distillation in all-glass systems may be required. Where possible, reagents are cleaned by extraction or solvent rinse.
4.2 Proper cleaning of glassware is extremely important, because glassware may not only contaminate the samples but may also remove the analytes of interest by adsorption on the glass surface.
4.2.1 Glassware should be rinsed with solvent and washed with a detergent solution as soon after use as is practical. Sonication of glassware containing a detergent solution for approximately 30 seconds may aid in cleaning. Glassware with removable parts, particularly separatory funnels with fluoropolymer stopcocks, must be disassembled prior to detergent washing.
4.2.2 After detergent washing, glassware should be rinsed immediately, first with methanol, then with hot tap water. The tap water rinse is followed by another methanol rinse, then acetone, and then methylene chloride.
4.2.3 Do not bake reusable glassware in an oven as a routine part of cleaning. Baking may be warranted after particularly dirty samples are encountered but should be minimized, as repeated baking of glassware may cause active sites on the glass surface that will irreversibly adsorb CDDs/CDFs.
4.2.4 Immediately prior to use, the Soxhlet apparatus should be pre-extracted with toluene for approximately three hours (see Sections 12.3.1 through 12.3.3). Separatory funnels should be shaken with methylene chloride/toluene (80/20 mixture) for two minutes, drained, and then shaken with pure methylene chloride for two minutes.
4.3 All materials used in the analysis shall be demonstrated to be free from interferences by running reference matrix method blanks initially and with each sample batch (samples started through the extraction process on a given 12-hour shift, to a maximum of 20 samples).
4.3.1 The reference matrix must simulate, as closely as possible, the sample matrix under test. Ideally, the reference matrix should not contain the CDDs/CDFs in detectable amounts, but should contain potential interferents in the concentrations expected to be found in the samples to be analyzed. For example, a reference sample of human adipose tissue containing pentachloronaphthalene can be used to exercise the cleanup systems when samples containing pentachloronaphthalene are expected.
4.3.2 When a reference matrix that simulates the sample matrix under test is not available, reagent water (Section 7.6.1) can be used to simulate water samples; playground sand (Section 7.6.2) or white quartz sand (Section 7.3.2) can be used to simulate soils; filter paper (Section 7.6.3) can be used to simulate papers and similar materials; and corn oil (Section 7.6.4) can be used to simulate tissues.
4.4 Interferences coextracted from samples will vary considerably from source to source, depending on the diversity of the site being sampled. Interfering compounds may be present at concentrations several orders of magnitude higher than the CDDs/CDFs. The most frequently encountered interferences are chlorinated biphenyls, methoxy biphenyls, hydroxydiphenyl ethers, benzylphenyl ethers, polynuclear aromatics, and pesticides. Because very low levels of CDDs/CDFs are measured by this method, the elimination of interferences is essential. The cleanup steps given in Section 13 can be used to reduce or eliminate these interferences and thereby permit reliable determination of the CDDs/CDFs at the levels shown in Table 2.
4.5 Each piece of reusable glassware should be numbered to associate that glassware with the processing of a particular sample. This will assist the laboratory in tracking possible sources of contamination for individual samples, identifying glassware associated with highly contaminated samples that may require extra cleaning, and determining when glassware should be discarded.
4.6 Cleanup of tissue - The natural lipid content of tissue can interfere in the analysis of tissue samples for the CDDs/CDFs. The lipid contents of different species and portions of tissue can vary widely. Lipids are soluble to varying degrees in various organic solvents and may be present in sufficient quantity to overwhelm the column chromatographic cleanup procedures used for cleanup of sample extracts. Lipids must be removed by the lipid removal procedures in Section 13.7, followed by alumina (Section 13.4) or Florisil (Section 13.8), and carbon (Section 13.5) as minimum additional cleanup steps. If chlorodiphenyl ethers are detected, as indicated by the presence of peaks at the exact m/z's monitored for these interferents, alumina and/or Florisil cleanup must be employed to eliminate these interferences.
5.0 Safety5.1 The toxicity or carcinogenicity of each compound or reagent used in this method has not been precisely determined; however, each chemical compound should be treated as a potential health hazard. Exposure to these compounds should be reduced to the lowest possible level.
5.1.1 The 2,3,7,8-TCDD isomer has been found to be acnegenic, carcinogenic, and teratogenic in laboratory animal studies. It is soluble in water to approximately 200 ppt and in organic solvents to 0.14%. On the basis of the available toxicological and physical properties of 2,3,7,8-TCDD, all of the CDDs/CDFs should be handled only by highly trained personnel thoroughly familiar with handling and cautionary procedures and the associated risks.
5.1.2 It is recommended that the laboratory purchase dilute standard solutions of the analytes in this method. However, if primary solutions are prepared, they shall be prepared in a hood, and a NIOSH/MESA approved toxic gas respirator shall be worn when high concentrations are handled.
5.2 The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material safety data sheets (MSDSs) should also be made available to all personnel involved in these analyses. It is also suggested that the laboratory perform personal hygiene monitoring of each analyst who uses this method and that the results of this monitoring be made available to the analyst. Additional information on laboratory safety can be found in References 10-13. The references and bibliography at the end of Reference 13 are particularly comprehensive in dealing with the general subject of laboratory safety.
5.3 The CDDs/CDFs and samples suspected to contain these compounds are handled using essentially the same techniques employed in handling radioactive or infectious materials. Well-ventilated, controlled access laboratories are required. Assistance in evaluating the health hazards of particular laboratory conditions may be obtained from certain consulting laboratories and from State Departments of Health or Labor, many of which have an industrial health service. The CDDs/CDFs are extremely toxic to laboratory animals. Each laboratory must develop a strict safety program for handling these compounds. The practices in References 2 and 14 are highly recommended.
5.3.1 Facility - When finely divided samples (dusts, soils, dry chemicals) are handled, all operations (including removal of samples from sample containers, weighing, transferring, and mixing) should be performed in a glove box demonstrated to be leak tight or in a fume hood demonstrated to have adequate air flow. Gross losses to the laboratory ventilation system must not be allowed. Handling of the dilute solutions normally used in analytical and animal work presents no inhalation hazards except in the case of an accident.
5.3.2 Protective equipment - Disposable plastic gloves, apron or lab coat, safety glasses or mask, and a glove box or fume hood adequate for radioactive work should be used. During analytical operations that may give rise to aerosols or dusts, personnel should wear respirators equipped with activated carbon filters. Eye protection equipment (preferably full face shields) must be worn while working with exposed samples or pure analytical standards. Latex gloves are commonly used to reduce exposure of the hands. When handling samples suspected or known to contain high concentrations of the CDDs/CDFs, an additional set of gloves can also be worn beneath the latex gloves.
5.3.3 Training - Workers must be trained in the proper method of removing contaminated gloves and clothing without contacting the exterior surfaces.
5.3.4 Personal hygiene - Hands and forearms should be washed thoroughly after each manipulation and before breaks (coffee, lunch, and shift).
5.3.5 Confinement - Isolated work areas posted with signs, segregated glassware and tools, and plastic absorbent paper on bench tops will aid in confining contamination.
5.3.6 Effluent vapors - The effluents of sample splitters from the gas chromatograph (GC) and from roughing pumps on the mass spectrometer (MS) should pass through either a column of activated charcoal or be bubbled through a trap containing oil or high-boiling alcohols to condense CDD/CDF vapors.
5.3.7 Waste Handling - Good technique includes minimizing contaminated waste. Plastic bag liners should be used in waste cans. Janitors and other personnel must be trained in the safe handling of waste.
5.3.8 Decontamination
5.3.8.1 Decontamination of personnel - Use any mild soap with plenty of scrubbing action.
5.3.8.2 Glassware, tools, and surfaces - Chlorothene NU Solvent is the least toxic solvent shown to be effective. Satisfactory cleaning may be accomplished by rinsing with Chlorothene, then washing with any detergent and water. If glassware is first rinsed with solvent, then the dish water may be disposed of in the sewer. Given the cost of disposal, it is prudent to minimize solvent wastes.
5.3.9 Laundry - Clothing known to be contaminated should be collected in plastic bags. Persons who convey the bags and launder the clothing should be advised of the hazard and trained in proper handling. The clothing may be put into a washer without contact if the launderer knows of the potential problem. The washer should be run through a cycle before being used again for other clothing.
5.3.10 Wipe tests - A useful method of determining cleanliness of work surfaces and tools is to wipe the surface with a piece of filter paper. Extraction and analysis by GC with an electron capture detector (ECD) can achieve a limit of detection of 0.1 µg per wipe; analysis using this method can achieve an even lower detection limit. Less than 0.1 µg per wipe indicates acceptable cleanliness; anything higher warrants further cleaning. More than 10 µg on a wipe constitutes an acute hazard and requires prompt cleaning before further use of the equipment or work space, and indicates that unacceptable work practices have been employed.
5.3.11 Table or wrist-action shaker - The use of a table or wrist-action shaker for extraction of tissues presents the possibility of breakage of the extraction bottle and spillage of acid and flammable organic solvent. A secondary containment system around the shaker is suggested to prevent the spread of acid and solvents in the event of such a breakage. The speed and intensity of shaking action should also be adjusted to minimize the possibility of breakage.
6.0 Apparatus and Materials Note:Brand names, suppliers, and part numbers are for illustration purposes only and no endorsement is implied. Equivalent performance may be achieved using apparatus and materials other than those specified here. Meeting the performance requirements of this method is the responsibility of the laboratory.
6.1 Sampling Equipment for Discrete or Composite Sampling
6.1.1 Sample bottles and caps
6.1.1.1 Liquid samples (waters, sludges and similar materials containing 5% solids or less) - Sample bottle, amber glass, 1.1 L minimum, with screw cap.
6.1.1.2 Solid samples (soils, sediments, sludges, paper pulps, filter cake, compost, and similar materials that contain more than 5% solids) - Sample bottle, wide mouth, amber glass, 500 mL minimum.
6.1.1.3 If amber bottles are not available, samples shall be protected from light.
6.1.1.4 Bottle caps - Threaded to fit sample bottles. Caps shall be lined with fluoropolymer.
6.1.1.5 Cleaning
6.1.1.5.1 Bottles are detergent water washed, then solvent rinsed before use.
6.1.1.5.2 Liners are detergent water washed, rinsed with reagent water (Section 7.6.1) followed by solvent, and baked at approximately 200 °C for a minimum of 1 hour prior to use.
6.1.2 Compositing equipment - Automatic or manual compositing system incorporating glass containers cleaned per bottle cleaning procedure above. Only glass or fluoropolymer tubing shall be used. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used in the pump only. Before use, the tubing shall be thoroughly rinsed with methanol, followed by repeated rinsing with reagent water to minimize sample contamination. An integrating flow meter is used to collect proportional composite samples.
6.2 Equipment for Glassware Cleaning - Laboratory sink with overhead fume hood.
6.3 Equipment for Sample Preparation
6.3.1 Laboratory fume hood of sufficient size to contain the sample preparation equipment listed below.
6.3.2 Glove box (optional).
6.3.3 Tissue homogenizer - VirTis Model 45 Macro homogenizer (American Scientific Products H-3515, or equivalent) with stainless steel Macro-shaft and Turbo-shear blade.
6.3.4 Meat grinder - Hobart, or equivalent, with 3-5 mm holes in inner plate.
6.3.5 Equipment for determining percent moisture
6.3.5.1 Oven - Capable of maintaining a temperature of 110 ±5 °C.
6.3.5.2 Dessicator.
6.3.6 Balances
6.3.6.1 Analytical - Capable of weighing 0.1 mg.
6.3.6.2 Top loading - Capable of weighing 10 mg.
6.4 Extraction Apparatus
6.4.1 Water samples
6.4.1.1 pH meter, with combination glass electrode.
6.4.1.2 pH paper, wide range (Hydrion Papers, or equivalent).
6.4.1.3 Graduated cylinder, 1 L capacity.
6.4.1.4 Liquid/liquid extraction - Separatory funnels, 250 mL, 500 mL, and 2000 mL, with fluoropolymer stopcocks.
6.4.1.5 Solid-phase extraction
6.4.1.5.1 One liter filtration apparatus, including glass funnel, glass frit support, clamp, adapter, stopper, filtration flask, and vacuum tubing (Figure 4). For wastewater samples, the apparatus should accept 90 or 144 mm disks. For drinking water or other samples containing low solids, smaller disks may be used.
6.4.1.5.2 Vacuum source capable of maintaining 25 in. Hg, equipped with shutoff valve and vacuum gauge.
6.4.1.5.3 Glass-fiber filter - Whatman GMF 150 (or equivalent), 1 micron pore size, to fit filtration apparatus in Section 6.4.1.5.1.
6.4.1.5.4 Solid-phase extraction disk containing octadecyl (C18) bonded silica uniformly enmeshed in an inert matrix - Fisher Scientific 14-378F (or equivalent), to fit filtration apparatus in Section 6.4.1.5.1.
6.4.2 Soxhlet/Dean-Stark (SDS) extractor (Figure 5) - For filters and solid/sludge samples.
6.4.2.1 Soxhlet - 50 mm ID, 200 mL capacity with 500 mL flask (Cal-Glass LG-6900, or equivalent, except substitute 500 mL round-bottom flask for 300 mL flat-bottom flask).
6.4.2.2 Thimble - 43 × 123 to fit Soxhlet (Cal-Glass LG-6901-122, or equivalent).
6.4.2.3 Moisture trap - Dean Stark or Barret with fluoropolymer stopcock, to fit Soxhlet.
6.4.2.4 Heating mantle - Hemispherical, to fit 500 mL round-bottom flask (Cal-Glass LG-8801-112, or equivalent).
6.4.2.5 Variable transformer - Powerstat (or equivalent), 110 volt, 10 amp.
6.4.3 Apparatus for extraction of tissue.
6.4.3.1 Bottle for extraction (if digestion/extraction using HCl is used)” 500-600 mL wide-mouth clear glass, with fluoropolymer-lined cap.
6.4.3.2 Bottle for back-extraction - 100-200 mL narrow-mouth clear glass with fluoropolymer-lined cap.
6.4.3.3 Mechanical shaker - Wrist-action or platform-type rotary shaker that produces vigorous agitation (Sybron Thermolyne Model LE “Big Bill” rotator/shaker, or equivalent).
6.4.3.4 Rack attached to shaker table to permit agitation of four to nine samples simultaneously.
6.4.4 Beakers - 400-500 mL.
6.4.5 Spatulas - Stainless steel.
6.5 Filtration Apparatus.
6.5.1 Pyrex glass wool - Solvent-extracted by SDS for three hours minimum.
Note:Baking glass wool may cause active sites that will irreversibly adsorb CDDs/CDFs.
6.5.2 Glass funnel - 125-250 mL.
6.5.3 Glass-fiber filter paper - Whatman GF/D (or equivalent), to fit glass funnel in Section 6.5.2.
6.5.4 Drying column - 15-20 mm ID Pyrex chromatographic column equipped with coarse-glass frit or glass-wool plug.
6.5.5 Buchner funnel - 15 cm.
6.5.6 Glass-fiber filter paper - to fit Buchner funnel in Section 6.5.5.
6.5.7 Filtration flasks - 1.5-2.0 L, with side arm.
6.5.8 Pressure filtration apparatus - Millipore YT30 142 HW, or equivalent.
6.6 Centrifuge Apparatus.
6.6.1 Centrifuge - Capable of rotating 500 mL centrifuge bottles or 15 mL centrifuge tubes at 5,000 rpm minimum.
6.6.2 Centrifuge bottles - 500 mL, with screw-caps, to fit centrifuge.
6.6.3 Centrifuge tubes - 12-15 mL, with screw-caps, to fit centrifuge.
6.7 Cleanup Apparatus.
6.7.1 Automated gel permeation chromatograph (Analytical Biochemical Labs, Inc, Columbia, MO, Model GPC Autoprep 1002, or equivalent).
6.7.1.1 Column - 600-700 mm long × 25 mm ID, packed with 70 g of
SX-3 Bio-beads (Bio-Rad Laboratories, Richmond, CA, or equivalent).6.7.1.2 Syringe - 10 mL, with Luer fitting.
6.7.1.3 Syringe filter holder - stainless steel, and glass-fiber or fluoropolymer filters (Gelman 4310, or equivalent).
6.7.1.4 UV detectors - 254 nm, preparative or semi-preparative flow cell (Isco, Inc., Type 6; Schmadzu, 5 mm path length; Beckman-Altex 152W, 8 µL micro-prep flow cell, 2 mm path; Pharmacia UV-1, 3 mm flow cell; LDC Milton-Roy UV-3, monitor #1203; or equivalent).
6.7.2 Reverse-phase high-performance liquid chromatograph.
6.7.2.1 Column oven and detector - Perkin-Elmer Model LC-65T (or equivalent) operated at 0.02 AUFS at 235 nm.
6.7.2.2 Injector - Rheodyne 7120 (or equivalent) with 50 µL sample loop.
6.7.2.3 Column - Two 6.2 mm × 250 mm Zorbax-ODS columns in series (DuPont Instruments Division, Wilmington, DE, or equivalent), operated at 50 °C with 2.0 mL/min methanol isocratic effluent.
6.7.2.4 Pump - Altex 110A (or equivalent).
6.7.3 Pipets.
6.7.3.1 Disposable, pasteur - 150 mm long × 5-mm ID (Fisher Scientific 13-678-6A, or equivalent).
6.7.3.2 Disposable, serological - 10 mL (6 mm ID).
6.7.4 Glass chromatographic columns.
6.7.4.1 150 mm long × 8-mm ID, (Kontes K-420155, or equivalent) with coarse-glass frit or glass-wool plug and 250 mL reservoir.
6.7.4.2 200 mm long × 15 mm ID, with coarse-glass frit or glass-wool plug and 250 mL reservoir.
6.7.4.3 300 mm long × 25 mm ID, with 300 mL reservoir and glass or fluoropolymer stopcock.
6.7.5 Stirring apparatus for batch silica cleanup of tissue extracts.
6.7.5.1 Mechanical stirrer - Corning Model 320, or equivalent.
6.7.5.2 Bottle - 500-600 mL wide-mouth clear glass.
6.7.6 Oven - For baking and storage of adsorbents, capable of maintaining a constant temperature (±5 °C) in the range of 105-250 °C.
6.8 Concentration Apparatus.
6.8.1 Rotary evaporator - Buchi/Brinkman-American Scientific No. E5045-10 or equivalent, equipped with a variable temperature water bath.
6.8.1.1 Vacuum source for rotary evaporator equipped with shutoff valve at the evaporator and vacuum gauge.
6.8.1.2 A recirculating water pump and chiller are recommended, as use of tap water for cooling the evaporator wastes large volumes of water and can lead to inconsistent performance as water temperatures and pressures vary.
6.8.1.3 Round-bottom flask - 100 mL and 500 mL or larger, with ground-glass fitting compatible with the rotary evaporator.
6.8.2 Kuderna-Danish (K-D) Concentrator.
6.8.2.1 Concentrator tube - 10 mL, graduated (Kontes K-570050-1025, or equivalent) with calibration verified. Ground-glass stopper (size 19/22 joint) is used to prevent evaporation of extracts.
6.8.2.2 Evaporation flask - 500 mL (Kontes K-570001-0500, or equivalent), attached to concentrator tube with springs (Kontes K-662750-0012 or equivalent).
6.8.2.3 Snyder column - Three-ball macro (Kontes K-503000-0232, or equivalent).
6.8.2.4 Boiling chips.
6.8.2.4.1 Glass or silicon carbide - Approximately 10/40 mesh, extracted with methylene chloride and baked at 450 °C for one hour minimum.
6.8.2.4.2 Fluoropolymer (optional) - Extracted with methylene chloride.
6.8.2.5 Water bath - Heated, with concentric ring cover, capable of maintaining a temperature within ±2 °C, installed in a fume hood.
6.8.3 Nitrogen blowdown apparatus - Equipped with water bath controlled in the range of 30-60 °C (N-Evap, Organomation Associates, Inc., South Berlin, MA, or equivalent), installed in a fume hood.
6.8.4 Sample vials.
6.8.4.1 Amber glass - 2-5 mL with fluoropolymer-lined screw-cap.
6.8.4.2 Glass - 0.3 mL, conical, with fluoropolymer-lined screw or crimp cap.
6.9 Gas Chromatograph - Shall have splitless or on-column injection port for capillary column, temperature program with isothermal hold, and shall meet all of the performance specifications in Section 10.
6.9.1 GC column for CDDs/CDFs and for isomer specificity for 2,3,7,8-TCDD - 60 ±5 m long × 0.32 ±0.02 mm ID; 0.25 µm 5% phenyl, 94% methyl, 1% vinyl silicone bonded-phase fused-silica capillary column (J&W DB-5, or equivalent).
6.9.2 GC column for isomer specificity for 2,3,7,8-TCDF - 30 ±5 m long × 0.32 ±0.02 mm ID; 0.25 µm bonded-phase fused-silica capillary column (J&W DB-225, or equivalent).
6.10 Mass Spectrometer - 28-40 eV electron impact ionization, shall be capable of repetitively selectively monitoring 12 exact m/z's minimum at high resolution (≥10,000) during a period of approximately one second, and shall meet all of the performance specifications in Section 10.
6.11 GC/MS Interface - The mass spectrometer (MS) shall be interfaced to the GC such that the end of the capillary column terminates within 1 cm of the ion source but does not intercept the electron or ion beams.
6.12 Data System - Capable of collecting, recording, and storing MS data.
7.0 Reagents and Standards7.1 pH Adjustment and Back-Extraction.
7.1.1 Potassium hydroxide - Dissolve 20 g reagent grade KOH in 100 mL reagent water.
7.1.2 Sulfuric acid - Reagent grade (specific gravity 1.84).
7.1.3 Hydrochloric acid - Reagent grade, 6N.
7.1.4 Sodium chloride - Reagent grade, prepare at 5% (w/v) solution in reagent water.
7.2 Solution Drying and Evaporation.
7.2.1 Solution drying - Sodium sulfate, reagent grade, granular, anhydrous (Baker 3375, or equivalent), rinsed with methylene chloride (20 mL/g), baked at 400 °C for one hour minimum, cooled in a dessicator, and stored in a pre-cleaned glass bottle with screw-cap that prevents moisture from entering. If, after heating, the sodium sulfate develops a noticeable grayish cast (due to the presence of carbon in the crystal matrix), that batch of reagent is not suitable for use and should be discarded. Extraction with methylene chloride (as opposed to simple rinsing) and baking at a lower temperature may produce sodium sulfate that is suitable for use.
7.2.2 Tissue drying - Sodium sulfate, reagent grade, powdered, treated and stored as above.
7.2.3 Prepurified nitrogen.
7.3 Extraction.
7.3.1 Solvents - Acetone, toluene, cyclohexane, hexane, methanol, methylene chloride, and nonane; distilled in glass, pesticide quality, lot-certified to be free of interferences.
7.3.2 White quartz sand, 60/70 mesh - For Soxhlet/Dean-Stark extraction (Aldrich Chemical, Cat. No. 27-437-9, or equivalent). Bake at 450 °C for four hours minimum.
7.4 GPC Calibration Solution - Prepare a solution containing 300 mg/mL corn oil, 15 mg/mL bis(2-ethylhexyl) phthalate, 1.4 mg/mL pentachlorophenol, 0.1 mg/mL perylene, and 0.5 mg/mL sulfur.
7.5 Adsorbents for Sample Cleanup.
7.5.1 Silica gel.
7.5.1.1 Activated silica gel - 100-200 mesh, Supelco 1-3651 (or equivalent), rinsed with methylene chloride, baked at 180 °C for a minimum of one hour, cooled in a dessicator, and stored in a precleaned glass bottle with screw-cap that prevents moisture from entering.
7.5.1.2 Acid silica gel (30% w/w) - Thoroughly mix 44.0 g of concentrated sulfuric acid with 100.0 g of activated silica gel in a clean container. Break up aggregates with a stirring rod until a uniform mixture is obtained. Store in a bottle with a fluoropolymer-lined screw-cap.
7.5.1.3 Basic silica gel - Thoroughly mix 30 g of 1N sodium hydroxide with 100 g of activated silica gel in a clean container. Break up aggregates with a stirring rod until a uniform mixture is obtained. Store in a bottle with a fluoropolymer-lined screw-cap.
7.5.1.4 Potassium silicate.
7.5.1.4.1 Dissolve 56 g of high purity potassium hydroxide (Aldrich, or equivalent) in 300 mL of methanol in a 750-1000 mL flat-bottom flask.
7.5.1.4.2 Add 100 g of silica gel and a stirring bar, and stir on a hot plate at 60-70 °C for one to two hours.
7.5.1.4.3 Decant the liquid and rinse the potassium silicate twice with 100 mL portions of methanol, followed by a single rinse with 100 mL of methylene chloride.
7.5.1.4.4 Spread the potassium silicate on solvent-rinsed aluminum foil and dry for two to four hours in a hood.
7.5.1.4.5 Activate overnight at 200-250 °C.
7.5.2 Alumina - Either one of two types of alumina, acid or basic, may be used in the cleanup of sample extracts, provided that the laboratory can meet the performance specifications for the recovery of labeled compounds described in Section 9.3. The same type of alumina must be used for all samples, including those used to demonstrate initial precision and recovery (Section 9.2) and ongoing precision and recovery (Section 15.5).
7.5.2.1 Acid alumina - Supelco 19996-6C (or equivalent). Activate by heating to 130 °C for a minimum of 12 hours.
7.5.2.2 Basic alumina - Supelco 19944-6C (or equivalent). Activate by heating to 600 °C for a minimum of 24 hours. Alternatively, activate by heating in a tube furnace at 650-700 °C under an air flow rate of approximately 400 cc/minute. Do not heat over 700 °C, as this can lead to reduced capacity for retaining the analytes. Store at 130 °C in a covered flask. Use within five days of baking.
7.5.3 Carbon.
7.5.3.1 Carbopak C - (Supelco 1-0258, or equivalent).
7.5.3.2 Celite 545 - (Supelco 2-0199, or equivalent).
7.5.3.3 Thoroughly mix 9.0 g Carbopak C and 41.0 g Celite 545 to produce an 18% w/w mixture. Activate the mixture at 130 °C for a minimum of six hours. Store in a dessicator.
7.5.4 Anthropogenic isolation column - Pack the column in Section 6.7.4.3 from bottom to top with the following:
7.5.4.1 2 g silica gel (Section 7.5.1.1).
7.5.4.2 2 g potassium silicate (Section 7.5.1.4).
7.5.4.3 2 g granular anhydrous sodium sulfate (Section 7.2.1).
7.5.4.4 10 g acid silica gel (Section 7.5.1.2).
7.5.4.5 2 g granular anhydrous sodium sulfate.
7.5.5 Florisil column.
7.5.5.1 Florisil - 60-100 mesh, Floridin Corp (or equivalent). Soxhlet extract in 500 g portions for 24 hours.
7.5.5.2 Insert a glass wool plug into the tapered end of a graduated serological pipet (Section 6.7.3.2). Pack with 1.5 g (approx 2 mL) of Florisil topped with approx 1 mL of sodium sulfate (Section 7.2.1) and a glass wool plug.
7.5.5.3 Activate in an oven at 130-150 °C for a minimum of 24 hours and cool for 30 minutes. Use within 90 minutes of cooling.
7.6 Reference Matrices - Matrices in which the CDDs/CDFs and interfering compounds are not detected by this method.
7.6.1 Reagent water - Bottled water purchased locally, or prepared by passage through activated carbon.
7.6.2 High-solids reference matrix - Playground sand or similar material. Prepared by extraction with methylene chloride and/or baking at 450 °C for a minimum of four hours.
7.6.3 Paper reference matrix - Glass-fiber filter, Gelman Type A, or equivalent. Cut paper to simulate the surface area of the paper sample being tested.
7.6.4 Tissue reference matrix - Corn or other vegetable oil. May be prepared by extraction with methylene chloride.
7.6.5 Other matrices - This method may be verified on any reference matrix by performing the tests given in Section 9.2. Ideally, the matrix should be free of the CDDs/CDFs, but in no case shall the background level of the CDDs/CDFs in the reference matrix exceed three times the minimum levels in Table 2. If low background levels of the CDDs/CDFs are present in the reference matrix, the spike level of the analytes used in Section 9.2 should be increased to provide a spike-to-background ratio in the range of 1:1 to 5:1 (Reference 15).
7.7 Standard Solutions - Purchased as solutions or mixtures with certification to their purity, concentration, and authenticity, or prepared from materials of known purity and composition. If the chemical purity is 98% or greater, the weight may be used without correction to compute the concentration of the standard. When not being used, standards are stored in the dark at room temperature in screw-capped vials with fluoropolymer-lined caps. A mark is placed on the vial at the level of the solution so that solvent loss by evaporation can be detected. If solvent loss has occurred, the solution should be replaced.
7.8 Stock Solutions.
7.8.1 Preparation - Prepare in nonane per the steps below or purchase as dilute solutions (Cambridge Isotope Laboratories (CIL), Woburn, MA, or equivalent). Observe the safety precautions in Section 5, and the recommendation in Section 5.1.2.
7.8.2 Dissolve an appropriate amount of assayed reference material in solvent. For example, weigh 1-2 mg of 2,3,7,8-TCDD to three significant figures in a 10 mL ground-glass-stoppered volumetric flask and fill to the mark with nonane. After the TCDD is completely dissolved, transfer the solution to a clean 15 mL vial with fluoropolymer-lined cap.
7.8.3 Stock standard solutions should be checked for signs of degradation prior to the preparation of calibration or performance test standards. Reference standards that can be used to determine the accuracy of calibration standards are available from CIL and may be available from other vendors.
7.9 PAR Stock Solution
7.9.1 All CDDs/CDFs - Using the solutions in Section 7.8, prepare the PAR stock solution to contain the CDDs/CDFs at the concentrations shown in Table 3. When diluted, the solution will become the PAR (Section 7.14).
7.9.2 If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, prepare the PAR stock solution to contain these compounds only.
7.10 Labeled-Compound Spiking Solution.
7.10.1 All CDDs/CDFs - From stock solutions, or from purchased mixtures, prepare this solution to contain the labeled compounds in nonane at the concentrations shown in Table 3. This solution is diluted with acetone prior to use (Section 7.10.3).
7.10.2 If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, prepare the labeled-compound solution to contain these compounds only. This solution is diluted with acetone prior to use (Section 7.10.3).
7.10.3 Dilute a sufficient volume of the labeled compound solution (Section 7.10.1 or 7.10.2) by a factor of 50 with acetone to prepare a diluted spiking solution. Each sample requires 1.0 mL of the diluted solution, but no more solution should be prepared than can be used in one day.
7.11 Cleanup Standard - Prepare 37Cl 4-2,3,7,8-TCDD in nonane at the concentration shown in Table 3. The cleanup standard is added to all extracts prior to cleanup to measure the efficiency of the cleanup process.
7.12 Internal Standard(s).
7.12.1 All CDDs/CDFs - Prepare the internal standard solution to contain 13C 12-1,2,3,4-TCDD and 13C 2-1,2,3,7,8,9-HxCDD in nonane at the concentration shown in Table 3.
7.12.2 If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, prepare the internal standard solution to contain 13C 12-1,2,3,4-TCDD only.
7.13 Calibration Standards (CS1 through CS5) - Combine the solutions in Sections 7.9 through 7.12 to produce the five calibration solutions shown in Table 4 in nonane. These solutions permit the relative response (labeled to native) and response factor to be measured as a function of concentration. The CS3 standard is used for calibration verification (VER). If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, combine the solutions appropriate to these compounds.
7.14 Precision and Recovery (PAR) Standard - Used for determination of initial (Section 9.2) and ongoing (Section 15.5) precision and recovery. Dilute 10 µL of the precision and recovery standard (Section 7.9.1 or 7.9.2) to 2.0 mL with acetone for each sample matrix for each sample batch. One mL each are required for the blank and OPR with each matrix in each batch.
7.15 GC Retention Time Window Defining Solution and Isomer Specificity Test Standard - Used to define the beginning and ending retention times for the dioxin and furan isomers and to demonstrate isomer specificity of the GC columns employed for determination of 2,3,7,8-TCDD and 2,3,7,8-TCDF. The standard must contain the compounds listed in Table 5 (CIL EDF - 4006, or equivalent), at a minimum. It is not necessary to monitor the window-defining compounds if only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined. In this case, an isomer-specificity test standard containing the most closely eluted isomers listed in Table 5 (CIL EDF-4033, or equivalent) may be used.
7.16 QC Check Sample - A QC Check Sample should be obtained from a source independent of the calibration standards. Ideally, this check sample would be a certified reference material containing the CDDs/CDFs in known concentrations in a sample matrix similar to the matrix under test.
7.17 Stability of Solutions - Standard solutions used for quantitative purposes (Sections 7.9 through 7.15) should be analyzed periodically, and should be assayed against reference standards (Section 7.8.3) before further use.
8.0 Sample Collection, Preservation, Storage, and Holding Times8.1 Collect samples in amber glass containers following conventional sampling practices (Reference 16). Aqueous samples that flow freely are collected in refrigerated bottles using automatic sampling equipment. Solid samples are collected as grab samples using wide-mouth jars.
8.2 Maintain aqueous samples in the dark at 0-4 °C from the time of collection until receipt at the laboratory. If residual chlorine is present in aqueous samples, add 80 mg sodium thiosulfate per liter of water. EPA Methods 330.4 and 330.5 may be used to measure residual chlorine (Reference 17). If sample pH is greater than 9, adjust to pH 7-9 with sulfuric acid.
Maintain solid, semi-solid, oily, and mixed-phase samples in the dark at <4 °C from the time of collection until receipt at the laboratory.
Store aqueous samples in the dark at 0-4 °C. Store solid, semi-solid, oily, mixed-phase, and tissue samples in the dark at <−10 °C.
8.3 Fish and Tissue Samples.
8.3.1 Fish may be cleaned, filleted, or processed in other ways in the field, such that the laboratory may expect to receive whole fish, fish fillets, or other tissues for analysis.
8.3.2 Fish collected in the field should be wrapped in aluminum foil, and must be maintained at a temperature less than 4 °C from the time of collection until receipt at the laboratory.
8.3.3 Samples must be frozen upon receipt at the laboratory and maintained in the dark at <−10 °C until prepared. Maintain unused sample in the dark at <−10 °C.
8.4 Holding Times.
8.4.1 There are no demonstrated maximum holding times associated with CDDs/CDFs in aqueous, solid, semi-solid, tissues, or other sample matrices. If stored in the dark at 0-4 °C and preserved as given above (if required), aqueous samples may be stored for up to one year. Similarly, if stored in the dark at <−10 °C, solid, semi-solid, multi-phase, and tissue samples may be stored for up to one year.
8.4.2 Store sample extracts in the dark at <−10 °C until analyzed. If stored in the dark at <−10 °C, sample extracts may be stored for up to one year.
9.0 Quality Assurance/Quality Control9.1 Each laboratory that uses this method is required to operate a formal quality assurance program (Reference 18). The minimum requirements of this program consist of an initial demonstration of laboratory capability, analysis of samples spiked with labeled compounds to evaluate and document data quality, and analysis of standards and blanks as tests of continued performance. Laboratory performance is compared to established performance criteria to determine if the results of analyses meet the performance characteristics of the method.
If the method is to be applied to sample matrix other than water (e.g., soils, filter cake, compost, tissue) the most appropriate alternate matrix (Sections 7.6.2 through 7.6.5) is substituted for the reagent water matrix (Section 7.6.1) in all performance tests.
9.1.1 The analyst shall make an initial demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 9.2.
9.1.2 In recognition of advances that are occurring in analytical technology, and to allow the analyst to overcome sample matrix interferences, the analyst is permitted certain options to improve separations or lower the costs of measurements. These options include alternate extraction, concentration, cleanup procedures, and changes in columns and detectors. Alternate determinative techniques, such as the substitution of spectroscopic or immuno-assay techniques, and changes that degrade method performance, are not allowed. If an analytical technique other than the techniques specified in this method is used, that technique must have a specificity equal to or better than the specificity of the techniques in this method for the analytes of interest.
9.1.2.1 Each time a modification is made to this method, the analyst is required to repeat the procedure in Section 9.2. If the detection limit of the method will be affected by the change, the laboratory is required to demonstrate that the MDL (40 CFR part 136, appendix B) is lower than one-third the regulatory compliance level or one-third the ML in this method, whichever is higher. If calibration will be affected by the change, the analyst must recalibrate the instrument per Section 10.
9.1.2.2 The laboratory is required to maintain records of modifications made to this method. These records include the following, at a minimum:
9.1.2.2.1 The names, titles, addresses, and telephone numbers of the analyst(s) who performed the analyses and modification, and of the quality control officer who witnessed and will verify the analyses and modifications.
9.1.2.2.2 A listing of pollutant(s) measured, by name and CAS Registry number.
9.1.2.2.3 A narrative stating reason(s) for the modifications.
9.1.2.2.4 Results from all quality control (QC) tests comparing the modified method to this method, including:
(a) Calibration (Section 10.5 through 10.7).
(b) Calibration verification (Section 15.3).
(c) Initial precision and recovery (Section 9.2).
(d) Labeled compound recovery (Section 9.3).
(e) Analysis of blanks (Section 9.5).
(f) Accuracy assessment (Section 9.4).
9.1.2.2.5 Data that will allow an independent reviewer to validate each determination by tracing the instrument output (peak height, area, or other signal) to the final result. These data are to include:
(a) Sample numbers and other identifiers.
(b) Extraction dates.
(c) Analysis dates and times.
(d) Analysis sequence/run chronology.
(e) Sample weight or volume (Section 11).
(f) Extract volume prior to each cleanup step (Section 13).
(g) Extract volume after each cleanup step (Section 13).
(h) Final extract volume prior to injection (Section 14).
(i) Injection volume (Section 14.3).
(j) Dilution data, differentiating between dilution of a sample or extract (Section 17.5).
(k) Instrument and operating conditions.
(l) Column (dimensions, liquid phase, solid support, film thickness, etc).
(m) Operating conditions (temperatures, temperature program, flow rates).
(n) Detector (type, operating conditions, etc).
(o) Chromatograms, printer tapes, and other recordings of raw data.
(p) Quantitation reports, data system outputs, and other data to link the raw data to the results reported.
9.1.3 Analyses of method blanks are required to demonstrate freedom from contamination (Section 4.3). The procedures and criteria for analysis of a method blank are described in Sections 9.5 and 15.6.
9.1.4 The laboratory shall spike all samples with labeled compounds to monitor method performance. This test is described in Section 9.3. When results of these spikes indicate atypical method performance for samples, the samples are diluted to bring method performance within acceptable limits. Procedures for dilution are given in Section 17.5.
9.1.5 The laboratory shall, on an ongoing basis, demonstrate through calibration verification and the analysis of the ongoing precision and recovery aliquot that the analytical system is in control. These procedures are described in Sections 15.1 through 15.5.
9.1.6 The laboratory shall maintain records to define the quality of data that is generated. Development of accuracy statements is described in Section 9.4.
9.2 Initial Precision and Recovery (IPR) - To establish the ability to generate acceptable precision and recovery, the analyst shall perform the following operations.
9.2.1 For low solids (aqueous) samples, extract, concentrate, and analyze four 1 L aliquots of reagent water spiked with the diluted labeled compound spiking solution (Section 7.10.3) and the precision and recovery standard (Section 7.14) according to the procedures in Sections 11 through 18. For an alternative sample matrix, four aliquots of the alternative reference matrix (Section 7.6) are used. All sample processing steps that are to be used for processing samples, including preparation (Section 11), extraction (Section 12), and cleanup (Section 13), shall be included in this test.
9.2.2 Using results of the set of four analyses, compute the average concentration (X) of the extracts in ng/mL and the standard deviation of the concentration (s) in ng/mL for each compound, by isotope dilution for CDDs/CDFs with a labeled analog, and by internal standard for 1,2,3,7,8,9-HxCDD, OCDF, and the labeled compounds.
9.2.3 For each CDD/CDF and labeled compound, compare s and X with the corresponding limits for initial precision and recovery in Table 6. If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, compare s and X with the corresponding limits for initial precision and recovery in Table 6a. If s and X for all compounds meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may begin. If, however, any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, system performance is unacceptable for that compound. Correct the problem and repeat the test (Section 9.2).
9.3 The laboratory shall spike all samples with the diluted labeled compound spiking solution (Section 7.10.3) to assess method performance on the sample matrix.
9.3.1 Analyze each sample according to the procedures in Sections 11 through 18.
9.3.2 Compute the percent recovery of the labeled compounds and the cleanup standard using the internal standard method (Section 17.2).
9.3.3 The recovery of each labeled compound must be within the limits in Table 7 when all 2,3,7,8-substituted CDDs/CDFs are determined, and within the limits in Table 7a when only 2,3,7,8-TCDD and 2,3,7,8-TCDF are determined. If the recovery of any compound falls outside of these limits, method performance is unacceptable for that compound in that sample. To overcome such difficulties, water samples are diluted and smaller amounts of soils, sludges, sediments, and other matrices are reanalyzed per Section 18.4.
9.4 Recovery of labeled compounds from samples should be assessed and records should be maintained.
9.4.1 After the analysis of five samples of a given matrix type (water, soil, sludge, pulp, etc.) for which the labeled compounds pass the tests in Section 9.3, compute the average percent recovery (R) and the standard deviation of the percent recovery (SR) for the labeled compounds only. Express the assessment as a percent recovery interval from R−2SR to R = 2SR for each matrix. For example, if R = 90% and SR = 10% for five analyses of pulp, the recovery interval is expressed as 70-110%.
9.4.2 Update the accuracy assessment for each labeled compound in each matrix on a regular basis (e.g., after each 5-10 new measurements).
9.5 Method Blanks - Reference matrix method blanks are analyzed to demonstrate freedom from contamination (Section 4.3).
9.5.1 Prepare, extract, clean up, and concentrate a method blank with each sample batch (samples of the same matrix started through the extraction process on the same 12-hour shift, to a maximum of 20 samples). The matrix for the method blank shall be similar to sample matrix for the batch, e.g., a 1 L reagent water blank (Section 7.6.1), high-solids reference matrix blank (Section 7.6.2), paper matrix blank (Section 7.6.3); tissue blank (Section 7.6.4) or alternative reference matrix blank (Section 7.6.5). Analyze the blank immediately after analysis of the OPR (Section 15.5) to demonstrate freedom from contamination.
9.5.2 If any 2,3,7,8-substituted CDD/CDF (Table 1) is found in the blank at greater than the minimum level (Table 2) or one-third the regulatory compliance level, whichever is greater; or if any potentially interfering compound is found in the blank at the minimum level for each level of chlorination given in Table 2 (assuming a response factor of 1 relative to the 13C12-1,2,3,4-TCDD internal standard for compounds not listed in Table 1), analysis of samples is halted until the blank associated with the sample batch shows no evidence of contamination at this level. All samples must be associated with an uncontaminated method blank before the results for those samples may be reported for regulatory compliance purposes.
9.6 QC Check Sample - Analyze the QC Check Sample (Section 7.16) periodically to assure the accuracy of calibration standards and the overall reliability of the analytical process. It is suggested that the QC Check Sample be analyzed at least quarterly.
9.7 The specifications contained in this method can be met if the apparatus used is calibrated properly and then maintained in a calibrated state. The standards used for calibration (Section 10), calibration verification (Section 15.3), and for initial (Section 9.2) and ongoing (Section 15.5) precision and recovery should be identical, so that the most precise results will be obtained. A GC/MS instrument will provide the most reproducible results if dedicated to the settings and conditions required for the analyses of CDDs/CDFs by this method.
9.8 Depending on specific program requirements, field replicates may be collected to determine the precision of the sampling technique, and spiked samples may be required to determine the accuracy of the analysis when the internal standard method is used.
10.0 Calibration10.1 Establish the operating conditions necessary to meet the minimum retention times for the internal standards in Section 10.2.4 and the relative retention times for the CDDs/CDFs in Table 2.
10.1.1 Suggested GC operating conditions:
Injector temperature: 270 °C Interface temperature: 290 °C Initial temperature: 200 °C Initial time: Two minutes Temperature program: 200-220 °C, at 5 °C/minute 220 °C for 16 minutes 220-235 °C, at 5 °C/minute 235 °C for seven minutes 235-330 °C, at 5 °C/minute Note:All portions of the column that connect the GC to the ion source shall remain at or above the interface temperature specified above during analysis to preclude condensation of less volatile compounds.
Optimize GC conditions for compound separation and sensitivity. Once optimized, the same GC conditions must be used for the analysis of all standards, blanks, IPR and OPR aliquots, and samples.
10.1.2 Mass spectrometer (MS) resolution - Obtain a selected ion current profile (SICP) of each analyte in Table 3 at the two exact m/z's specified in Table 8 and at ≥10,000 resolving power by injecting an authentic standard of the CDDs/CDFs either singly or as part of a mixture in which there is no interference between closely eluted components.
10.1.2.1 The analysis time for CDDs/CDFs may exceed the long-term mass stability of the mass spectrometer. Because the instrument is operated in the high-resolution mode, mass drifts of a few ppm (e.g., 5 ppm in mass) can have serious adverse effects on instrument performance. Therefore, a mass-drift correction is mandatory and a lock-mass m/z from PFK is used for drift correction. The lock-mass m/z is dependent on the exact m/z's monitored within each descriptor, as shown in Table 8. The level of PFK metered into the HRMS during analyses should be adjusted so that the amplitude of the most intense selected lock-mass m/z signal (regardless of the descriptor number) does not exceed 10% of the full-scale deflection for a given set of detector parameters. Under those conditions, sensitivity changes that might occur during the analysis can be more effectively monitored.
Note:Excessive PFK (or any other reference substance) may cause noise problems and contamination of the ion source necessitating increased frequency of source cleaning.
10.1.2.2 If the HRMS has the capability to monitor resolution during the analysis, it is acceptable to terminate the analysis when the resolution falls below 10,000 to save reanalysis time.
10.1.2.3 Using a PFK molecular leak, tune the instrument to meet the minimum required resolving power of 10,000 (10% valley) at m/z 304.9824 (PFK) or any other reference signal close to m/z 304 (from TCDF). For each descriptor (Table 8), monitor and record the resolution and exact m/z's of three to five reference peaks covering the mass range of the descriptor. The resolution must be greater than or equal to 10,000, and the deviation between the exact m/z and the theoretical m/z (Table 8) for each exact m/z monitored must be less than 5 ppm.
10.2 Ion Abundance Ratios, Minimum Levels, Signal-to-Noise Ratios, and Absolute Retention Times - Choose an injection volume of either 1 µL or 2 µL, consistent with the capability of the HRGC/HRMS instrument. Inject a 1 µL or 2 µL aliquot of the CS1 calibration solution (Table 4) using the GC conditions from Section 10.1.1. If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, the operating conditions and specifications below apply to analysis of those compounds only.
10.2.1 Measure the SICP areas for each analyte, and compute the ion abundance ratios at the exact m/z's specified in Table 8. Compare the computed ratio to the theoretical ratio given in Table 9.
10.2.1.1 The exact m/z's to be monitored in each descriptor are shown in Table 8. Each group or descriptor shall be monitored in succession as a function of GC retention time to ensure that all CDDs/CDFs are detected. Additional m/z's may be monitored in each descriptor, and the m/z's may be divided among more than the five descriptors listed in Table 8, provided that the laboratory is able to monitor the m/z's of all the CDDs/CDFs that may elute from the GC in a given retention-time window. If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, the descriptors may be modified to include only the exact m/z's for the tetra-and penta-isomers, the diphenyl ethers, and the lock m/z's.
10.2.1.2 The mass spectrometer shall be operated in a mass-drift correction mode, using perfluorokerosene (PFK) to provide lock m/z's. The lock-mass for each group of m/z's is shown in Table 8. Each lock mass shall be monitored and shall not vary by more than ±20% throughout its respective retention time window. Variations of the lock mass by more than 20% indicate the presence of coeluting interferences that may significantly reduce the sensitivity of the mass spectrometer. Reinjection of another aliquot of the sample extract will not resolve the problem. Additional cleanup of the extract may be required to remove the interferences.
10.2.2 All CDDs/CDFs and labeled compounds in the CS1 standard shall be within the QC limits in Table 9 for their respective ion abundance ratios; otherwise, the mass spectrometer shall be adjusted and this test repeated until the m/z ratios fall within the limits specified. If the adjustment alters the resolution of the mass spectrometer, resolution shall be verified (Section 10.1.2) prior to repeat of the test.
10.2.3 Verify that the HRGC/HRMS instrument meets the minimum levels in Table 2. The peaks representing the CDDs/CDFs and labeled compounds in the CS1 calibration standard must have signal-to-noise ratios (S/N) greater than or equal to 10.0. Otherwise, the mass spectrometer shall be adjusted and this test repeated until the minimum levels in Table 2 are met.
10.2.4 The absolute retention time of 13C12-1,2,3,4-TCDD (Section 7.12) shall exceed 25.0 minutes on the DB-5 column, and the retention time of 13C12-1,2,3,4-TCDD shall exceed 15.0 minutes on the DB-225 column; otherwise, the GC temperature program shall be adjusted and this test repeated until the above-stated minimum retention time criteria are met.
2010.3 Retention-Time Windows - Analyze the window defining mixtures (Section 7.15) using the optimized temperature program in Section 10.1. Table 5 gives the elution order (first/last) of the window-defining compounds. If 2,3,7,8-TCDD and 2,3,7,8-TCDF only are to be analyzed, this test is not required.10.4 Isomer Specificity.
10.4.1 Analyze the isomer specificity test standards (Section 7.15) using the procedure in Section 14 and the optimized conditions for sample analysis (Section 10.1.1).
10.4.2 Compute the percent valley between the GC peaks that elute most closely to the 2,3,7,8-TCDD and TCDF isomers, on their respective columns, per Figures 6 and 7.
10.4.3 Verify that the height of the valley between the most closely eluted isomers and the 2,3,7,8-substituted isomers is less than 25% (computed as 100 x/y in Figures 6 and 7). If the valley exceeds 25%, adjust the analytical conditions and repeat the test or replace the GC column and recalibrate (Sections 10.1.2 through 10.7).
10.5 Calibration by Isotope Dilution - Isotope dilution calibration is used for the 15 2,3,7,8-substituted CDDs/CDFs for which labeled compounds are added to samples prior to extraction. The reference compound for each CDD/CDF compound is shown in Table 2.
10.5.1 A calibration curve encompassing the concentration range is prepared for each compound to be determined. The relative response (RR) (labeled to native) vs. concentration in standard solutions is plotted or computed using a linear regression. Relative response is determined according to the procedures described below. Five calibration points are employed.
10.5.2 The response of each CDD/CDF relative to its labeled analog is determined using the area responses of both the primary and secondary exact m/z's specified in Table 8, for each calibration standard, as follows:
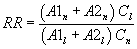 where:
A1n and A2n = The areas of the primary and secondary m/z's for the
CDD/CDF. A1l and A2l = The areas of the primary and secondary m/z's
for the labeled compound. Cl = The concentration of the labeled
compound in the calibration standard (Table 4). Cn = The
concentration of the native compound in the calibration standard
(Table 4).
where:
A1n and A2n = The areas of the primary and secondary m/z's for the
CDD/CDF. A1l and A2l = The areas of the primary and secondary m/z's
for the labeled compound. Cl = The concentration of the labeled
compound in the calibration standard (Table 4). Cn = The
concentration of the native compound in the calibration standard
(Table 4).
10.5.3 To calibrate the analytical system by isotope dilution, inject a volume of calibration standards CS1 through CS5 (Section 7.13 and Table 4) identical to the volume chosen in Section 10.2, using the procedure in Section 14 and the conditions in Section 10.1.1 and Table 2. Compute the relative response (RR) at each concentration.
10.5.4 Linearity - If the relative response for any compound is constant (less than 20% coefficient of variation) over the five-point calibration range, an averaged relative response may be used for that compound; otherwise, the complete calibration curve for that compound shall be used over the five-point calibration range.
10.6 Calibration by Internal Standard - The internal standard method is applied to determination of 1,2,3,7,8,9-HxCDD (Section 17.1.2), OCDF (Section 17.1.1), the non 2,3,7,8-substituted compounds, and to the determination of labeled compounds for intralaboratory statistics (Sections 9.4 and 15.5.4).
10.6.1 Response factors - Calibration requires the determination of response factors (RF) defined by the following equation:
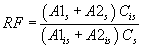 where:
A1s and A2s = The areas of the primary and secondary m/z's for the
CDD/CDF. A1is and A2is = The areas of the primary and secondary
m/z's for the internal standard. Cis = The concentration of the
internal standard (Table 4). Cs = The concentration of the compound
in the calibration standard (Table 4). Note:
where:
A1s and A2s = The areas of the primary and secondary m/z's for the
CDD/CDF. A1is and A2is = The areas of the primary and secondary
m/z's for the internal standard. Cis = The concentration of the
internal standard (Table 4). Cs = The concentration of the compound
in the calibration standard (Table 4). Note:
There is only one m/z for 37Cl4-2,3,7,8-TCDD. See Table 8.
10.6.2 To calibrate the analytical system by internal standard, inject 1.0 µL or 2.0 µL of calibration standards CS1 through CS5 (Section 7.13 and Table 4) using the procedure in Section 14 and the conditions in Section 10.1.1 and Table 2. Compute the response factor (RF) at each concentration.
10.6.3 Linearity - If the response factor (RF) for any compound is constant (less than 35% coefficient of variation) over the five-point calibration range, an averaged response factor may be used for that compound; otherwise, the complete calibration curve for that compound shall be used over the five-point range.
10.7 Combined Calibration - By using calibration solutions (Section 7.13 and Table 4) containing the CDDs/CDFs and labeled compounds and the internal standards, a single set of analyses can be used to produce calibration curves for the isotope dilution and internal standard methods. These curves are verified each shift (Section 15.3) by analyzing the calibration verification standard (VER, Table 4). Recalibration is required if any of the calibration verification criteria (Section 15.3) cannot be met.
10.8 Data Storage - MS data shall be collected, recorded, and stored.
10.8.1 Data acquisition - The signal at each exact m/z shall be collected repetitively throughout the monitoring period and stored on a mass storage device.
10.8.2 Response factors and multipoint calibrations - The data system shall be used to record and maintain lists of response factors (response ratios for isotope dilution) and multipoint calibration curves. Computations of relative standard deviation (coefficient of variation) shall be used to test calibration linearity. Statistics on initial performance (Section 9.2) and ongoing performance (Section 15.5) should be computed and maintained, either on the instrument data system, or on a separate computer system.
11.0 Sample Preparation11.1 Sample preparation involves modifying the physical form of the sample so that the CDDs/CDFs can be extracted efficiently. In general, the samples must be in a liquid form or in the form of finely divided solids in order for efficient extraction to take place. Table 10 lists the phases and suggested quantities for extraction of various sample matrices.
For samples known or expected to contain high levels of the CDDs/CDFs, the smallest sample size representative of the entire sample should be used (see Section 17.5).
For all samples, the blank and IPR/OPR aliquots must be processed through the same steps as the sample to check for contamination and losses in the preparation processes.
11.1.1 For samples that contain particles, percent solids and particle size are determined using the procedures in Sections 11.2 and 11.3, respectively.
11.1.2 Aqueous samples - Because CDDs/CDFs may be bound to suspended particles, the preparation of aqueous samples is dependent on the solids content of the sample.
11.1.2.1 Aqueous samples visibly absent particles are prepared per Section 11.4 and extracted directly using the separatory funnel or SPE techniques in Sections 12.1 or 12.2, respectively.
11.1.2.2 Aqueous samples containing visible particles and containing one percent suspended solids or less are prepared using the procedure in Section 11.4. After preparation, the sample is extracted directly using the SPE technique in 12.2 or filtered per Section 11.4.3. After filtration, the particles and filter are extracted using the SDS procedure in Section 12.3 and the filtrate is extracted using the separatory funnel procedure in Section 12.1.
11.1.2.3 For aqueous samples containing greater than one percent solids, a sample aliquot sufficient to provide 10 g of dry solids is used, as described in Section 11.5.
11.1.3 Solid samples are prepared using the procedure described in Section 11.5 followed by extraction via the SDS procedure in Section 12.3.
11.1.4 Multiphase samples - The phase(s) containing the CDDs/CDFs is separated from the non-CDD/CDF phase using pressure filtration and centrifugation, as described in Section 11.6. The CDDs/CDFs will be in the organic phase in a multiphase sample in which an organic phase exists.
11.1.5 Procedures for grinding, homogenization, and blending of various sample phases are given in Section 11.7.
11.1.6 Tissue samples - Preparation procedures for fish and other tissues are given in Section 11.8.
11.2 Determination of Percent Suspended Solids.
Note:This aliquot is used for determining the solids content of the sample, not for determination of CDDs/CDFs.
11.2.1 Aqueous liquids and multi-phase samples consisting of mainly an aqueous phase.
11.2.1.1 Dessicate and weigh a GF/D filter (Section 6.5.3) to three significant figures.
11.2.1.2 Filter 10.0 ±0.02 mL of well-mixed sample through the filter.
11.2.1.3 Dry the filter a minimum of 12 hours at 110 ±5 °C and cool in a dessicator.
11.2.1.4 Calculate percent solids as follows:
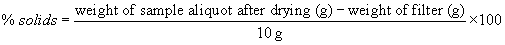
11.2.2 Non-aqueous liquids, solids, semi-solid samples, and multi-phase samples in which the main phase is not aqueous; but not tissues.
11.2.2.1 Weigh 5-10 g of sample to three significant figures in a tared beaker.
11.2.2.2 Dry a minimum of 12 hours at 110 ±5 °C, and cool in a dessicator.
11.2.2.3 Calculate percent solids as follows:
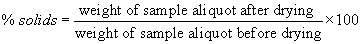
11.3 Determination of Particle Size.
11.3.1 Spread the dried sample from Section 11.2.2.2 on a piece of filter paper or aluminum foil in a fume hood or glove box.
11.3.2 Estimate the size of the particles in the sample. If the size of the largest particles is greater than 1 mm, the particle size must be reduced to 1 mm or less prior to extraction using the procedures in Section 11.7.
11.4 Preparation of Aqueous Samples Containing 1% Suspended Solids or Less.
11.4.1 Aqueous samples visibly absent particles are prepared per the procedure below and extracted directly using the separatory funnel or SPE techniques in Sections 12.1 or 12.2, respectively. Aqueous samples containing visible particles and one percent suspended solids or less are prepared using the procedure below and extracted using either the SPE technique in Section 12.2 or further prepared using the filtration procedure in Section 11.4.3. The filtration procedure is followed by SDS extraction of the filter and particles (Section 12.3) and separatory funnel extraction of the filtrate (Section 12.1). The SPE procedure is followed by SDS extraction of the filter and disk.
11.4.2 Preparation of sample and QC aliquots.
11.4.2.1 Mark the original level of the sample on the sample bottle for reference. Weigh the sample plus bottle to ±1.
11.4.2.2 Spike 1.0 mL of the diluted labeled-compound spiking solution (Section 7.10.3) into the sample bottle. Cap the bottle and mix the sample by careful shaking. Allow the sample to equilibrate for one to two hours, with occasional shaking.
11.4.2.3 For each sample or sample batch (to a maximum of 20 samples) to be extracted during the same 12-hour shift, place two 1.0 L aliquots of reagent water in clean sample bottles or flasks.
11.4.2.4 Spike 1.0 mL of the diluted labeled-compound spiking solution (Section 7.10.3) into both reagent water aliquots. One of these aliquots will serve as the method blank.
11.4.2.5 Spike 1.0 mL of the PAR standard (Section 7.14) into the remaining reagent water aliquot. This aliquot will serve as the OPR (Section 15.5).
11.4.2.6 If SPE is to be used, add 5 mL of methanol to the sample, cap and shake the sample to mix thoroughly, and proceed to Section 12.2 for extraction. If SPE is not to be used, and the sample is visibly absent particles, proceed to Section 12.1 for extraction. If SPE is not to be used and the sample contains visible particles, proceed to the following section for filtration of particles.
11.4.3 Filtration of particles.
11.4.3.1 Assemble a Buchner funnel (Section 6.5.5) on top of a clean filtration flask. Apply vacuum to the flask, and pour the entire contents of the sample bottle through a glass-fiber filter (Section 6.5.6) in the Buchner funnel, swirling the sample remaining in the bottle to suspend any particles.
11.4.3.2 Rinse the sample bottle twice with approximately 5 mL portions of reagent water to transfer any remaining particles onto the filter.
11.4.3.3 Rinse any particles off the sides of the Buchner funnel with small quantities of reagent water.
11.4.3.4 Weigh the empty sample bottle to ±1 g. Determine the weight of the sample by difference. Save the bottle for further use.
11.4.3.5 Extract the filtrate using the separatory funnel procedure in Section 12.1.
11.4.3.6 Extract the filter containing the particles using the SDS procedure in Section 12.3.
11.5 Preparation of Samples Containing Greater Than 1% Solids.
11.5.1 Weigh a well-mixed aliquot of each sample (of the same matrix type) sufficient to provide 10 g of dry solids (based on the solids determination in Section 11.2) into a clean beaker or glass jar.
11.5.2 Spike 1.0 mL of the diluted labeled compound spiking solution (Section 7.10.3) into the sample.
11.5.3 For each sample or sample batch (to a maximum of 20 samples) to be extracted during the same 12-hour shift, weigh two 10 g aliquots of the appropriate reference matrix (Section 7.6) into clean beakers or glass jars.
11.5.4 Spike 1.0 mL of the diluted labeled compound spiking solution (Section 7.10.3) into each reference matrix aliquot. One aliquot will serve as the method blank. Spike 1.0 mL of the PAR standard (Section 7.14) into the other reference matrix aliquot. This aliquot will serve as the OPR (Section 15.5).
11.5.5 Stir or tumble and equilibrate the aliquots for one to two hours.
11.5.6 Decant excess water. If necessary to remove water, filter the sample through a glass-fiber filter and discard the aqueous liquid.
11.5.7 If particles >1mm are present in the sample (as determined in Section 11.3.2), spread the sample on clean aluminum foil in a hood. After the sample is dry, grind to reduce the particle size (Section 11.7).
11.5.8 Extract the sample and QC aliquots using the SDS procedure in Section 12.3.
11.6 Multiphase Samples.
11.6.1 Using the percent solids determined in Section 11.2.1 or 11.2.2, determine the volume of sample that will provide 10 g of solids, up to 1 L of sample.
11.6.2 Pressure filter the amount of sample determined in Section 11.6.1 through Whatman GF/D glass-fiber filter paper (Section 6.5.3). Pressure filter the blank and OPR aliquots through GF/D papers also. If necessary to separate the phases and/or settle the solids, centrifuge these aliquots prior to filtration.
11.6.3 Discard any aqueous phase (if present). Remove any non-aqueous liquid present and reserve the maximum amount filtered from the sample (Section 11.6.1) or 10 g, whichever is less, for combination with the solid phase (Section 12.3.5).
11.6.4 If particles >1mm are present in the sample (as determined in Section 11.3.2) and the sample is capable of being dried, spread the sample and QC aliquots on clean aluminum foil in a hood. After the aliquots are dry or if the sample cannot be dried, reduce the particle size using the procedures in Section 11.7 and extract the reduced particles using the SDS procedure in Section 12.3. If particles >1mm are not present, extract the particles and filter in the sample and QC aliquots directly using the SDS procedure in Section 12.3.
11.7 Sample grinding, homogenization, or blending - Samples with particle sizes greater than 1 mm (as determined in Section 11.3.2) are subjected to grinding, homogenization, or blending. The method of reducing particle size to less than 1 mm is matrix-dependent. In general, hard particles can be reduced by grinding with a mortar and pestle. Softer particles can be reduced by grinding in a Wiley mill or meat grinder, by homogenization, or in a blender.
11.7.1 Each size-reducing preparation procedure on each matrix shall be verified by running the tests in Section 9.2 before the procedure is employed routinely.
11.7.2 The grinding, homogenization, or blending procedures shall be carried out in a glove box or fume hood to prevent particles from contaminating the work environment.
11.7.3 Grinding - Certain papers and pulps, slurries, and amorphous solids can be ground in a Wiley mill or heavy duty meat grinder. In some cases, reducing the temperature of the sample to freezing or to dry ice or liquid nitrogen temperatures can aid in the grinding process. Grind the sample aliquots from Section 11.5.7 or 11.6.4 in a clean grinder. Do not allow the sample temperature to exceed 50 °C. Grind the blank and reference matrix aliquots using a clean grinder.
11.7.4 Homogenization or blending - Particles that are not ground effectively, or particles greater than 1 mm in size after grinding, can often be reduced in size by high speed homogenization or blending. Homogenize and/or blend the particles or filter from Section 11.5.7 or 11.6.4 for the sample, blank, and OPR aliquots.
11.7.5 Extract the aliquots using the SDS procedure in Section 12.3.
11.8 Fish and Other Tissues - Prior to processing tissue samples, the laboratory must determine the exact tissue to be analyzed. Common requests for analysis of fish tissue include whole fish - skin on, whole fish - skin removed, edible fish fillets (filleted in the field or by the laboratory), specific organs, and other portions. Once the appropriate tissue has been determined, the sample must be homogenized.
11.8.1 Homogenization.
11.8.1.1 Samples are homogenized while still frozen, where practical. If the laboratory must dissect the whole fish to obtain the appropriate tissue for analysis, the unused tissues may be rapidly refrozen and stored in a clean glass jar for subsequent use.
11.8.1.2 Each analysis requires 10 g of tissue (wet weight). Therefore, the laboratory should homogenize at least 20 g of tissue to allow for re-extraction of a second aliquot of the same homogenized sample, if re-analysis is required. When whole fish analysis is necessary, the entire fish is homogenized.
11.8.1.3 Homogenize the sample in a tissue homogenizer (Section 6.3.3) or grind in a meat grinder (Section 6.3.4). Cut tissue too large to feed into the grinder into smaller pieces. To assure homogeneity, grind three times.
11.8.1.4 Transfer approximately 10 g (wet weight) of homogenized tissue to a clean, tared, 400-500 mL beaker. For the alternate HCl digestion/extraction, transfer the tissue to a clean, tared 500-600 mL wide-mouth bottle. Record the weight to the nearest 10 mg.
11.8.1.5 Transfer the remaining homogenized tissue to a clean jar with a fluoropolymer-lined lid. Seal the jar and store the tissue at <−10 °C. Return any tissue that was not homogenized to its original container and store at <−10 °C.
11.8.2 QC aliquots.
11.8.2.1 Prepare a method blank by adding approximately 10 g of the oily liquid reference matrix (Section 7.6.4) to a 400-500 mL beaker. For the alternate HCl digestion/extraction, add the reference matrix to a 500-600 mL wide-mouth bottle. Record the weight to the nearest 10 mg.
11.8.2.2 Prepare a precision and recovery aliquot by adding approximately 10 g of the oily liquid reference matrix (Section 7.6.4) to a separate 400-500 mL beaker or wide-mouth bottle, depending on the extraction procedure to be used. Record the weight to the nearest 10 mg. If the initial precision and recovery test is to be performed, use four aliquots; if the ongoing precision and recovery test is to be performed, use a single aliquot.
11.8.3 Spiking
11.8.3.1 Spike 1.0 mL of the labeled compound spiking solution (Section 7.10.3) into the sample, blank, and OPR aliquot.
11.8.3.2 Spike 1.0 mL of the PAR standard (Section 7.14) into the OPR aliquot.
11.8.4 Extract the aliquots using the procedures in Section 12.4.
12.0 Extraction and ConcentrationExtraction procedures include separatory funnel (Section 12.1) and solid phase (Section 12.2) for aqueous liquids; Soxhlet/Dean-Stark (Section 12.3) for solids, filters, and SPE disks; and Soxhlet extraction (Section 12.4.1) and HCl digestion (Section 12.4.2) for tissues. Acid/base back-extraction (Section 12.5) is used for initial cleanup of extracts.
Macro-concentration procedures include rotary evaporation (Section 12.6.1), heating mantle (Section 12.6.2), and Kuderna-Danish (K-D) evaporation (Section 12.6.3). Micro-concentration uses nitrogen blowdown (Section 12.7).
12.1 Separatory funnel extraction of filtrates and of aqueous samples visibly absent particles.
12.1.1 Pour the spiked sample (Section 11.4.2.2) or filtrate (Section 11.4.3.5) into a 2 L separatory funnel. Rinse the bottle or flask twice with 5 mL of reagent water and add these rinses to the separatory funnel.
12.1.2 Add 60 mL methylene chloride to the empty sample bottle (Section 12.1.1), seal, and shake 60 seconds to rinse the inner surface. Transfer the solvent to the separatory funnel, and extract the sample by shaking the funnel for two minutes with periodic venting. Allow the organic layer to separate from the aqueous phase for a minimum of 10 minutes. If an emulsion forms and is more than one-third the volume of the solvent layer, employ mechanical techniques to complete the phase separation (see note below). Drain the methylene chloride extract through a solvent-rinsed glass funnel approximately one-half full of granular anhydrous sodium sulfate (Section 7.2.1) supported on clean glass-fiber paper into a solvent-rinsed concentration device (Section 12.6).
Note:If an emulsion forms, the analyst must employ mechanical techniques to complete the phase separation. The optimum technique depends upon the sample, but may include stirring, filtration through glass wool, use of phase separation paper, centrifugation, use of an ultrasonic bath with ice, addition of NaCl, or other physical methods. Alternatively, solid-phase or other extraction techniques may be used to prevent emulsion formation. Any alternative technique is acceptable so long as the requirements in Section 9 are met.
Experience with aqueous samples high in dissolved organic materials (e.g., paper mill effluents) has shown that acidification of the sample prior to extraction may reduce the formation of emulsions. Paper industry methods suggest that the addition of up to 400 mL of ethanol to a 1 L effluent sample may also reduce emulsion formation. However, studies by EPA suggest that the effect may be a result of sample dilution, and that the addition of reagent water may serve the same function. Mechanical techniques may still be necessary to complete the phase separation. If either acidification or addition of ethanol is utilized, the laboratory must perform the startup tests described in Section 9.2 using the same techniques.
12.1.3 Extract the water sample two more times with 60 mL portions of methylene chloride. Drain each portion through the sodium sulfate into the concentrator. After the third extraction, rinse the separatory funnel with at least 20 mL of methylene chloride, and drain this rinse through the sodium sulfate into the concentrator. Repeat this rinse at least twice. Set aside the funnel with sodium sulfate if the extract is to be combined with the extract from the particles.
12.1.4 Concentrate the extract using one of the macro-concentration procedures in Section 12.6.
12.1.4.1 If the extract is from a sample visibly absent particles (Section 11.1.2.1), adjust the final volume of the concentrated extract to approximately 10 mL with hexane, transfer to a 250 mL separatory funnel, and back-extract using the procedure in Section 12.5.
12.1.4.2 If the extract is from the aqueous filtrate (Section 11.4.3.5), set aside the concentration apparatus for addition of the SDS extract from the particles (Section 12.3.9.1.2).
12.2 SPE of Samples Containing Less Than 1% Solids (References 19-20).
12.2.1 Disk preparation.
12.2.1.1 Place an SPE disk on the base of the filter holder (Figure 4) and wet with toluene. While holding a GMF 150 filter above the SPE disk with tweezers, wet the filter with toluene and lay the filter on the SPE disk, making sure that air is not trapped between the filter and disk. Clamp the filter and SPE disk between the 1 L glass reservoir and the vacuum filtration flask.
12.2.1.2 Rinse the sides of the filtration flask with approx 15 mL of toluene using a squeeze bottle or syringe. Apply vacuum momentarily until a few drops appear at the drip tip. Release the vacuum and allow the filter/disk to soak for approx one minute. Apply vacuum and draw all of the toluene through the filter/disk. Repeat the wash step with approx 15 mL of acetone and allow the filter/disk to air dry.
12.2.1.3 Re-wet the filter/disk with approximately 15 mL of methanol, allowing the filter/disk to soak for approximately one minute. Pull the methanol through the filter/disk using the vacuum, but retain a layer of methanol approximately 1 mm thick on the filter. Do not allow the disk to go dry from this point until the end of the extraction.
12.2.1.4 Rinse the filter/disk with two 50-mL portions of reagent water by adding the water to the reservoir and pulling most through, leaving a layer of water on the surface of the filter.
12.2.2 Extraction.
12.2.2.1 Pour the spiked sample (Section 11.4.2.2), blank (Section 11.4.2.4), or IPR/OPR aliquot (Section 11.4.2.5) into the reservoir and turn on the vacuum to begin the extraction. Adjust the vacuum to complete the extraction in no less than 10 minutes. For samples containing a high concentration of particles (suspended solids), filtration times may be eight hours or longer.
12.2.2.2 Before all of the sample has been pulled through the filter/disk, rinse the sample bottle with approximately 50 mL of reagent water to remove any solids, and pour into the reservoir. Pull through the filter/disk. Use additional reagent water rinses until all visible solids are removed.
12.2.2.3 Before all of the sample and rinses have been pulled through the filter/disk, rinse the sides of the reservoir with small portions of reagent water.
12.2.2.4 Allow the filter/disk to dry, then remove the filter and disk and place in a glass Petri dish. Extract the filter and disk per Section 12.3.
12.3 SDS Extraction of Samples Containing Particles, and of Filters and/or Disks.
12.3.1 Charge a clean extraction thimble (Section 6.4.2.2) with 5.0 g of 100/200 mesh silica (Section 7.5.1.1) topped with 100 g of quartz sand (Section 7.3.2).
Note:Do not disturb the silica layer throughout the extraction process.
12.3.2 Place the thimble in a clean extractor. Place 30-40 mL of toluene in the receiver and 200-250 mL of toluene in the flask.
12.3.3 Pre-extract the glassware by heating the flask until the toluene is boiling. When properly adjusted, one to two drops of toluene will fall per second from the condenser tip into the receiver. Extract the apparatus for a minimum of three hours.
12.3.4 After pre-extraction, cool and disassemble the apparatus. Rinse the thimble with toluene and allow to air dry.
12.3.5 Load the wet sample, filter, and/or disk from Section 11.4.3.6, 11.5.8, 11.6.4, 11.7.3, 11.7.4, or 12.2.2.4 and any nonaqueous liquid from Section 11.6.3 into the thimble and manually mix into the sand layer with a clean metal spatula, carefully breaking up any large lumps of sample.
12.3.6 Reassemble the pre-extracted SDS apparatus, and add a fresh charge of toluene to the receiver and reflux flask. Apply power to the heating mantle to begin refluxing. Adjust the reflux rate to match the rate of percolation through the sand and silica beds until water removal lessens the restriction to toluene flow. Frequently check the apparatus for foaming during the first two hours of extraction. If foaming occurs, reduce the reflux rate until foaming subsides.
12.3.7 Drain the water from the receiver at one to two hours and eight to nine hours, or sooner if the receiver fills with water. Reflux the sample for a total of 16-24 hours. Cool and disassemble the apparatus. Record the total volume of water collected.
12.3.8 Remove the distilling flask. Drain the water from the Dean-Stark receiver and add any toluene in the receiver to the extract in the flask.
12.3.9 Concentrate the extract using one of the macro-concentration procedures in Section 12.6 per the following:
12.3.9.1 Extracts from the particles in an aqueous sample containing less than 1% solids (Section 11.4.3.6).
12.3.9.1.1 Concentrate the extract to approximately 5 mL using the rotary evaporator or heating mantle procedures in Section 12.6.1 or 12.6.2.
12.3.9.1.2 Quantitatively transfer the extract through the sodium sulfate (Section 12.1.3) into the apparatus that was set aside (Section 12.1.4.2) and reconcentrate to the level of the toluene.
12.3.9.1.3 Adjust to approximately 10 mL with hexane, transfer to a 250 mL separatory funnel, and proceed with back-extraction (Section 12.5).
12.3.9.2 Extracts from particles (Sections 11.5 through 11.6) or from the SPE filter and disk (Section 12.2.2.4) - Concentrate to approximately 10 mL using the rotary evaporator or heating mantle (Section 12.6.1 or 12.6.2), transfer to a 250 mL separatory funnel, and proceed with back-extraction (Section 12.5).
12.4 Extraction of Tissue - Two procedures are provided for tissue extraction.
12.4.1 Soxhlet extraction (Reference 21).
12.4.1.1 Add 30-40 g of powdered anhydrous sodium sulfate to each of the beakers (Section 11.8.4) and mix thoroughly. Cover the beakers with aluminum foil and allow to equilibrate for 12-24 hours. Remix prior to extraction to prevent clumping.
12.4.1.2 Assemble and pre-extract the Soxhlet apparatus per Sections 12.3.1 through 12.3.4, except use the methylene chloride:hexane (1:1) mixture for the pre-extraction and rinsing and omit the quartz sand. The Dean-Stark moisture trap may also be omitted, if desired.
12.4.1.3 Reassemble the pre-extracted Soxhlet apparatus and add a fresh charge of methylene chloride:hexane to the reflux flask.
12.4.1.4 Transfer the sample/sodium sulfate mixture (Section 12.4.1.1) to the Soxhlet thimble, and install the thimble in the Soxhlet apparatus.
12.4.1.5 Rinse the beaker with several portions of solvent mixture and add to the thimble. Fill the thimble/receiver with solvent. Extract for 18-24 hours.
12.4.1.6 After extraction, cool and disassemble the apparatus.
12.4.1.7 Quantitatively transfer the extract to a macro-concentration device (Section 12.6), and concentrate to near dryness. Set aside the concentration apparatus for re-use.
12.4.1.8 Complete the removal of the solvent using the nitrogen blowdown procedure (Section 12.7) and a water bath temperature of 60 °C. Weigh the receiver, record the weight, and return the receiver to the blowdown apparatus, concentrating the residue until a constant weight is obtained.
12.4.1.9 Percent lipid determination - The lipid content is determined by extraction of tissue with the same solvent system (methylene chloride:hexane) that was used in EPA's National Dioxin Study (Reference 22) so that lipid contents are consistent with that study.
12.4.1.9.1 Redissolve the residue in the receiver in hexane and spike 1.0 mL of the cleanup standard (Section 7.11) into the solution.
12.4.1.9.2 Transfer the residue/hexane to the anthropogenic isolation column (Section 13.7.1) or bottle for the acidified silica gel batch cleanup (Section 13.7.2), retaining the boiling chips in the concentration apparatus. Use several rinses to assure that all material is transferred. If necessary, sonicate or heat the receiver slightly to assure that all material is re-dissolved. Allow the receiver to dry. Weigh the receiver and boiling chips.
12.4.1.9.3 Calculate the lipid content to the nearest three significant figures as follows:

12.4.1.9.4 It is not necessary to determine the lipid content of the blank, IPR, or OPR aliquots.
12.4.2 HCl digestion/extraction and concentration (References 23-26).
12.4.2.1 Add 200 mL of 6 N HCl and 200 mL of methylene chloride:hexane (1:1) to the sample and QC aliquots (Section 11.8.4).
12.4.2.2 Cap and shake each bottle one to three times. Loosen the cap in a hood to vent excess pressure. Shake each bottle for 10-30 seconds and vent.
12.4.2.3 Tightly cap and place on shaker. Adjust the shaker action and speed so that the acid, solvent, and tissue are in constant motion. However, take care to avoid such violent action that the bottle may be dislodged from the shaker. Shake for 12-24 hours.
12.4.2.4 After digestion, remove the bottles from the shaker. Allow the bottles to stand so that the solvent and acid layers separate.
12.4.2.5 Decant the solvent through a glass funnel with glass-fiber filter (Sections 6.5.2 through 6.5.3) containing approximately 10 g of granular anhydrous sodium sulfate (Section 7.2.1) into a macro-concentration apparatus (Section 12.6). Rinse the contents of the bottle with two 25 mL portions of hexane and pour through the sodium sulfate into the apparatus.
12.4.2.6 Concentrate the solvent to near dryness using a macro-concentration procedure (Section 12.6).
12.4.2.7 Complete the removal of the solvent using the nitrogen blowdown apparatus (Section 12.7) and a water bath temperature of 60 °C. Weigh the receiver, record the weight, and return the receiver to the blowdown apparatus, concentrating the residue until a constant weight is obtained.
12.4.2.8 Percent lipid determination - The lipid content is determined in the same solvent system [methylene chloride:hexane (1:1)] that was used in EPA's National Dioxin Study (Reference 22) so that lipid contents are consistent with that study.
12.4.2.8.1 Redissolve the residue in the receiver in hexane and spike 1.0 mL of the cleanup standard (Section 7.11) into the solution.
12.4.2.8.2 Transfer the residue/hexane to the narrow-mouth 100-200 mL bottle retaining the boiling chips in the receiver. Use several rinses to assure that all material is transferred, to a maximum hexane volume of approximately 70 mL. Allow the receiver to dry. Weigh the receiver and boiling chips.
12.4.2.8.3 Calculate the percent lipid per Section 12.4.1.9.3. It is not necessary to determine the lipid content of the blank, IPR, or OPR aliquots.
12.4.2.9 Clean up the extract per Section 13.7.3.
12.5 Back-Extraction with Base and Acid.
12.5.1 Spike 1.0 mL of the cleanup standard (Section 7.11) into the separatory funnels containing the sample and QC extracts from Section 12.1.4.1, 12.3.9.1.3, or 12.3.9.2.
12.5.2 Partition the extract against 50 mL of potassium hydroxide solution (Section 7.1.1). Shake for two minutes with periodic venting into a hood. Remove and discard the aqueous layer. Repeat the base washing until no color is visible in the aqueous layer, to a maximum of four washings. Minimize contact time between the extract and the base to prevent degradation of the CDDs/CDFs. Stronger potassium hydroxide solutions may be employed for back-extraction, provided that the laboratory meets the specifications for labeled compound recovery and demonstrates acceptable performance using the procedure in Section 9.2.
12.5.3 Partition the extract against 50 mL of sodium chloride solution (Section 7.1.4) in the same way as with base. Discard the aqueous layer.
12.5.4 Partition the extract against 50 mL of sulfuric acid (Section 7.1.2) in the same way as with base. Repeat the acid washing until no color is visible in the aqueous layer, to a maximum of four washings.
12.5.5 Repeat the partitioning against sodium chloride solution and discard the aqueous layer.
12.5.6 Pour each extract through a drying column containing 7-10 cm of granular anhydrous sodium sulfate (Section 7.2.1). Rinse the separatory funnel with 30-50 mL of solvent, and pour through the drying column. Collect each extract in a round-bottom flask. Re-concentrate the sample and QC aliquots per Sections 12.6 through 12.7, and clean up the samples and QC aliquots per Section 13.
12.6 Macro-Concentration - Extracts in toluene are concentrated using a rotary evaporator or a heating mantle; extracts in methylene chloride or hexane are concentrated using a rotary evaporator, heating mantle, or Kuderna-Danish apparatus.
12.6.1 Rotary evaporation - Concentrate the extracts in separate round-bottom flasks.
12.6.1.1 Assemble the rotary evaporator according to manufacturer's instructions, and warm the water bath to 45 °C. On a daily basis, preclean the rotary evaporator by concentrating 100 mL of clean extraction solvent through the system. Archive both the concentrated solvent and the solvent in the catch flask for a contamination check if necessary. Between samples, three 2-3 mL aliquots of solvent should be rinsed down the feed tube into a waste beaker.
12.6.1.2 Attach the round-bottom flask containing the sample extract to the rotary evaporator. Slowly apply vacuum to the system, and begin rotating the sample flask.
12.6.1.3 Lower the flask into the water bath, and adjust the speed of rotation and the temperature as required to complete concentration in 15-20 minutes. At the proper rate of concentration, the flow of solvent into the receiving flask will be steady, but no bumping or visible boiling of the extract will occur.
Note:If the rate of concentration is too fast, analyte loss may occur.
12.6.1.4 When the liquid in the concentration flask has reached an apparent volume of approximately 2 mL, remove the flask from the water bath and stop the rotation. Slowly and carefully admit air into the system. Be sure not to open the valve so quickly that the sample is blown out of the flask. Rinse the feed tube with approximately 2 mL of solvent.
12.6.1.5 Proceed to Section 12.6.4 for preparation for back-extraction or micro-concentration and solvent exchange.
12.6.2 Heating mantle - Concentrate the extracts in separate round-bottom flasks.
12.6.2.1 Add one or two clean boiling chips to the round-bottom flask, and attach a three-ball macro Snyder column. Prewet the column by adding approximately 1 mL of solvent through the top. Place the round-bottom flask in a heating mantle, and apply heat as required to complete the concentration in 15-20 minutes. At the proper rate of distillation, the balls of the column will actively chatter, but the chambers will not flood.
12.6.2.2 When the liquid has reached an apparent volume of approximately 10 mL, remove the round-bottom flask from the heating mantle and allow the solvent to drain and cool for at least 10 minutes. Remove the Snyder column and rinse the glass joint into the receiver with small portions of solvent.
12.6.2.3 Proceed to Section 12.6.4 for preparation for back-extraction or micro-concentration and solvent exchange.
12.6.3 Kuderna-Danish (K-D) - Concentrate the extracts in separate 500 mL K-D flasks equipped with 10 mL concentrator tubes. The K-D technique is used for solvents such as methylene chloride and hexane. Toluene is difficult to concentrate using the K-D technique unless a water bath fed by a steam generator is used.
12.6.3.1 Add one to two clean boiling chips to the receiver. Attach a three-ball macro Snyder column. Prewet the column by adding approximately 1 mL of solvent through the top. Place the K-D apparatus in a hot water bath so that the entire lower rounded surface of the flask is bathed with steam.
12.6.3.2 Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15-20 minutes. At the proper rate of distillation, the balls of the column will actively chatter but the chambers will not flood.
12.6.3.3 When the liquid has reached an apparent volume of 1 mL, remove the K-D apparatus from the bath and allow the solvent to drain and cool for at least 10 minutes. Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1-2 mL of solvent. A 5 mL syringe is recommended for this operation.
12.6.3.4 Remove the three-ball Snyder column, add a fresh boiling chip, and attach a two-ball micro Snyder column to the concentrator tube. Prewet the column by adding approximately 0.5 mL of solvent through the top. Place the apparatus in the hot water bath.
12.6.3.5 Adjust the vertical position and the water temperature as required to complete the concentration in 5-10 minutes. At the proper rate of distillation, the balls of the column will actively chatter but the chambers will not flood.
12.6.3.6 When the liquid reaches an apparent volume of 0.5 mL, remove the apparatus from the water bath and allow to drain and cool for at least 10 minutes.
12.6.3.7 Proceed to 12.6.4 for preparation for back-extraction or micro-concentration and solvent exchange.
12.6.4 Preparation for back-extraction or micro-concentration and solvent exchange.
12.6.4.1 For back-extraction (Section 12.5), transfer the extract to a 250 mL separatory funnel. Rinse the concentration vessel with small portions of hexane, adjust the hexane volume in the separatory funnel to 10-20 mL, and proceed to back-extraction (Section 12.5).
12.6.4.2 For determination of the weight of residue in the extract, or for clean-up procedures other than back-extraction, transfer the extract to a blowdown vial using two to three rinses of solvent. Proceed with micro-concentration and solvent exchange (Section 12.7).
12.7 Micro-Concentration and Solvent Exchange.
12.7.1 Extracts to be subjected to GPC or HPLC cleanup are exchanged into methylene chloride. Extracts to be cleaned up using silica gel, alumina, carbon, and/or Florisil are exchanged into hexane.
12.7.2 Transfer the vial containing the sample extract to a nitrogen blowdown device. Adjust the flow of nitrogen so that the surface of the solvent is just visibly disturbed.
Note:A large vortex in the solvent may cause analyte loss.
12.7.3 Lower the vial into a 45 °C water bath and continue concentrating.
12.7.3.1 If the extract is to be concentrated to dryness for weight determination (Sections 12.4.1.8, 12.4.2.7, and 13.7.1.4), blow dry until a constant weight is obtained.
12.7.3.2 If the extract is to be concentrated for injection into the GC/MS or the solvent is to be exchanged for extract cleanup, proceed as follows:
12.7.4 When the volume of the liquid is approximately 100 L, add 2-3 mL of the desired solvent (methylene chloride for GPC and HPLC, or hexane for the other cleanups) and continue concentration to approximately 100 µL. Repeat the addition of solvent and concentrate once more.
12.7.5 If the extract is to be cleaned up by GPC, adjust the volume of the extract to 5.0 mL with methylene chloride. If the extract is to be cleaned up by HPLC, further concentrate the extract to 30 µL. Proceed with GPC or HPLC cleanup (Section 13.2 or 13.6, respectively).
12.7.6 If the extract is to be cleaned up by column chromatography (alumina, silica gel, Carbopak/Celite, or Florisil), bring the final volume to 1.0 mL with hexane. Proceed with column cleanups (Sections 13.3 through 13.5 and 13.8).
12.7.7 If the extract is to be concentrated for injection into the GC/MS (Section 14), quantitatively transfer the extract to a 0.3 mL conical vial for final concentration, rinsing the larger vial with hexane and adding the rinse to the conical vial. Reduce the volume to approximately 100 µL. Add 10 µL of nonane to the vial, and evaporate the solvent to the level of the nonane. Seal the vial and label with the sample number. Store in the dark at room temperature until ready for GC/MS analysis. If GC/MS analysis will not be performed on the same day, store the vial at <−10 °C.
13.0 Extract Cleanup13.1 Cleanup may not be necessary for relatively clean samples (e.g., treated effluents, groundwater, drinking water). If particular circumstances require the use of a cleanup procedure, the analyst may use any or all of the procedures below or any other appropriate procedure. Before using a cleanup procedure, the analyst must demonstrate that the requirements of Section 9.2 can be met using the cleanup procedure. If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, the cleanup procedures may be optimized for isolation of these two compounds.
13.1.1 Gel permeation chromatography (Section 13.2) removes high molecular weight interferences that cause GC column performance to degrade. It should be used for all soil and sediment extracts and may be used for water extracts that are expected to contain high molecular weight organic compounds (e.g., polymeric materials, humic acids).
13.1.2 Acid, neutral, and basic silica gel (Section 13.3), alumina (Section 13.4), and Florisil (Section 13.8) are used to remove nonpolar and polar interferences. Alumina and Florisil are used to remove chlorodiphenyl ethers.
13.1.3 Carbopak/Celite (Section 13.5) is used to remove nonpolar interferences.
13.1.4 HPLC (Section 13.6) is used to provide specificity for the 2,3,7,8-substituted and other CDD and CDF isomers.
13.1.5 The anthropogenic isolation column (Section 13.7.1), acidified silica gel batch adsorption procedure (Section 13.7.2), and sulfuric acid and base back-extraction (Section 13.7.3) are used for removal of lipids from tissue samples.
13.2 Gel Permeation Chromatography (GPC).
13.2.1 Column packing.
13.2.1.1 Place 70-75 g of SX-3 Bio-beads (Section 6.7.1.1) in a 400-500 mL beaker.
13.2.1.2 Cover the beads with methylene chloride and allow to swell overnight (a minimum of 12 hours).
13.2.1.3 Transfer the swelled beads to the column (Section 6.7.1.1) and pump solvent through the column, from bottom to top, at 4.5-5.5 mL/minute prior to connecting the column to the detector.
13.2.1.4 After purging the column with solvent for one to two hours, adjust the column head pressure to 7-10 psig and purge for four to five hours to remove air. Maintain a head pressure of 7-10 psig. Connect the column to the detector (Section 6.7.1.4).
13.2.2 Column calibration.
13.2.2.1 Load 5 mL of the calibration solution (Section 7.4) into the sample loop.
13.2.2.2 Inject the calibration solution and record the signal from the detector. The elution pattern will be corn oil, bis(2-ethyl hexyl)phthalate, pentachlorophenol, perylene, and sulfur.
13.2.2.3 Set the “dump time” to allow >85% removal of the corn oil and >85% collection of the phthalate.
13.2.2.4 Set the “collect time” to the peak minimum between perylene and sulfur.
13.2.2.5 Verify the calibration with the calibration solution after every 20 extracts. Calibration is verified if the recovery of the pentachlorophenol is greater than 85%. If calibration is not verified, the system shall be recalibrated using the calibration solution, and the previous 20 samples shall be re-extracted and cleaned up using the calibrated GPC system.
13.2.3 Extract cleanup - GPC requires that the column not be overloaded. The column specified in this method is designed to handle a maximum of 0.5 g of high molecular weight material in a 5 mL extract. If the extract is known or expected to contain more than 0.5 g, the extract is split into aliquots for GPC, and the aliquots are combined after elution from the column. The residue content of the extract may be obtained gravimetrically by evaporating the solvent from a 50 µL aliquot.
13.2.3.1 Filter the extract or load through the filter holder (Section 6.7.1.3) to remove the particles. Load the 5.0 mL extract onto the column.
13.2.3.2 Elute the extract using the calibration data determined in Section 13.2.2. Collect the eluate in a clean 400-500 mL beaker.
13.2.3.3 Rinse the sample loading tube thoroughly with methylene chloride between extracts to prepare for the next sample.
13.2.3.4 If a particularly dirty extract is encountered, a 5.0 mL methylene chloride blank shall be run through the system to check for carry-over.
13.2.3.5 Concentrate the eluate per Sections 12.6 and 12.7 for further cleanup or injection into the GC/MS.
13.3 Silica Gel Cleanup.
13.3.1 Place a glass-wool plug in a 15 mm ID chromatography column (Section 6.7.4.2). Pack the column bottom to top with: 1 g silica gel (Section 7.5.1.1), 4 g basic silica gel (Section 7.5.1.3), 1 g silica gel, 8 g acid silica gel (Section 7.5.1.2), 2 g silica gel, and 4 g granular anhydrous sodium sulfate (Section 7.2.1). Tap the column to settle the adsorbents.
13.3.2 Pre-elute the column with 50-100 mL of hexane. Close the stopcock when the hexane is within 1 mm of the sodium sulfate. Discard the eluate. Check the column for channeling. If channeling is present, discard the column and prepare another.
13.3.3 Apply the concentrated extract to the column. Open the stopcock until the extract is within 1 mm of the sodium sulfate.
13.3.4 Rinse the receiver twice with 1 mL portions of hexane, and apply separately to the column. Elute the CDDs/CDFs with 100 mL hexane, and collect the eluate.
13.3.5 Concentrate the eluate per Sections 12.6 and 12.7 for further cleanup or injection into the HPLC or GC/MS.
13.3.6 For extracts of samples known to contain large quantities of other organic compounds (such as paper mill effluents), it may be advisable to increase the capacity of the silica gel column. This may be accomplished by increasing the strengths of the acid and basic silica gels. The acid silica gel (Section 7.5.1.2) may be increased in strength to as much as 44% w/w (7.9 g sulfuric acid added to 10 g silica gel). The basic silica gel (Section 7.5.1.3) may be increased in strength to as much as 33% w/w (50 mL 1N NaOH added to 100 g silica gel), or the potassium silicate (Section 7.5.1.4) may be used.
Note:The use of stronger acid silica gel (44% w/w) may lead to charring of organic compounds in some extracts. The charred material may retain some of the analytes and lead to lower recoveries of CDDs/CDFs. Increasing the strengths of the acid and basic silica gel may also require different volumes of hexane than those specified above to elute the analytes off the column. Therefore, the performance of the method after such modifications must be verified by the procedure in Section 9.2.
13.4 Alumina Cleanup.
13.4.1 Place a glass-wool plug in a 15 mm ID chromatography column (Section 6.7.4.2).
13.4.2 If using acid alumina, pack the column by adding 6 g acid alumina (Section 7.5.2.1). If using basic alumina, substitute 6 g basic alumina (Section 7.5.2.2). Tap the column to settle the adsorbents.
13.4.3 Pre-elute the column with 50-100 mL of hexane. Close the stopcock when the hexane is within 1 mm of the alumina.
13.4.4 Discard the eluate. Check the column for channeling. If channeling is present, discard the column and prepare another.
13.4.5 Apply the concentrated extract to the column. Open the stopcock until the extract is within 1 mm of the alumina.
13.4.6 Rinse the receiver twice with 1 mL portions of hexane and apply separately to the column. Elute the interfering compounds with 100 mL hexane and discard the eluate.
13.4.7 The choice of eluting solvents will depend on the choice of alumina (acid or basic) made in Section 13.4.2.
13.4.7.1 If using acid alumina, elute the CDDs/CDFs from the column with 20 mL methylene chloride:hexane (20:80 v/v). Collect the eluate.
13.4.7.2 If using basic alumina, elute the CDDs/CDFs from the column with 20 mL methylene chloride:hexane (50:50 v/v). Collect the eluate.
13.4.8 Concentrate the eluate per Sections 12.6 and 12.7 for further cleanup or injection into the HPLC or GC/MS.
13.5 Carbon Column.
13.5.1 Cut both ends from a 10 mL disposable serological pipet (Section 6.7.3.2) to produce a 10 cm column. Fire-polish both ends and flare both ends if desired. Insert a glass-wool plug at one end, and pack the column with 0.55 g of Carbopak/Celite (Section 7.5.3.3) to form an adsorbent bed approximately 2 cm long. Insert a glass-wool plug on top of the bed to hold the adsorbent in place.
13.5.2 Pre-elute the column with 5 mL of toluene followed by 2 mL of methylene chloride: methanol:toluene (15:4:1 v/v), 1 mL of methylene chloride:cyclohexane (1:1 v/v), and 5 mL of hexane. If the flow rate of eluate exceeds 0.5 mL/minute, discard the column.
13.5.3 When the solvent is within 1 mm of the column packing, apply the sample extract to the column. Rinse the sample container twice with 1 mL portions of hexane and apply separately to the column. Apply 2 mL of hexane to complete the transfer.
13.5.4 Elute the interfering compounds with two 3 mL portions of hexane, 2 mL of methylene chloride:cyclohexane (1:1 v/v), and 2 mL of methylene chloride:methanol:toluene (15:4:1 v/v). Discard the eluate.
13.5.5 Invert the column, and elute the CDDs/CDFs with 20 mL of toluene. If carbon particles are present in the eluate, filter through glass-fiber filter paper.
13.5.6 Concentrate the eluate per Sections 12.6 and 12.7 for further cleanup or injection into the HPLC or GC/MS.
13.6 HPLC (Reference 6).
13.6.1 Column calibration.
13.6.1.1 Prepare a calibration standard containing the 2,3,7,8-substituted isomers and/or other isomers of interest at a concentration of approximately 500 pg/µL in methylene chloride.
13.6.1.2 Inject 30 µL of the calibration solution into the HPLC and record the signal from the detector. Collect the eluant for reuse. The elution order will be the tetra- through octa-isomers.
13.6.1.3 Establish the collection time for the tetra-isomers and for the other isomers of interest. Following calibration, flush the injection system with copious quantities of methylene chloride, including a minimum of five 50 µL injections while the detector is monitored, to ensure that residual CDDs/CDFs are removed from the system.
13.6.1.4 Verify the calibration with the calibration solution after every 20 extracts. Calibration is verified if the recovery of the CDDs/CDFs from the calibration standard (Section 13.6.1.1) is 75-125% compared to the calibration (Section 13.6.1.2). If calibration is not verified, the system shall be recalibrated using the calibration solution, and the previous 20 samples shall be re-extracted and cleaned up using the calibrated system.
13.6.2 Extract cleanup - HPLC requires that the column not be overloaded. The column specified in this method is designed to handle a maximum of 30 µL of extract. If the extract cannot be concentrated to less than 30 µL, it is split into fractions and the fractions are combined after elution from the column.
13.6.2.1 Rinse the sides of the vial twice with 30 µL of methylene chloride and reduce to 30 µL with the evaporation apparatus (Section 12.7).
13.6.2.2 Inject the 30 µL extract into the HPLC.
13.6.2.3 Elute the extract using the calibration data determined in Section 13.6.1. Collect the fraction(s) in a clean 20 mL concentrator tube containing 5 mL of hexane:acetone (1:1 v/v).
13.6.2.4 If an extract containing greater than 100 ng/mL of total CDD or CDF is encountered, a 30 µL methylene chloride blank shall be run through the system to check for carry-over.
13.6.2.5 Concentrate the eluate per Section 12.7 for injection into the GC/MS.
13.7 Cleanup of Tissue Lipids - Lipids are removed from the Soxhlet extract using either the anthropogenic isolation column (Section 13.7.1) or acidified silica gel (Section 13.7.2), or are removed from the HCl digested extract using sulfuric acid and base back-extraction (Section 13.7.3).
13.7.1 Anthropogenic isolation column (References 22 and 27) - Used for removal of lipids from the Soxhlet/SDS extraction (Section 12.4.1).
13.7.1.1 Prepare the column as given in Section 7.5.4.
13.7.1.2 Pre-elute the column with 100 mL of hexane. Drain the hexane layer to the top of the column, but do not expose the sodium sulfate.
13.7.1.3 Load the sample and rinses (Section 12.4.1.9.2) onto the column by draining each portion to the top of the bed. Elute the CDDs/CDFs from the column into the apparatus used for concentration (Section 12.4.1.7) using 200 mL of hexane.
13.7.1.4 Concentrate the cleaned up extract (Sections 12.6 through 12.7) to constant weight per Section 12.7.3.1. If more than 500 mg of material remains, repeat the cleanup using a fresh anthropogenic isolation column.
13.7.1.5 Redissolve the extract in a solvent suitable for the additional cleanups to be used (Sections 13.2 through 13.6 and 13.8).
13.7.1.6 Spike 1.0 mL of the cleanup standard (Section 7.11) into the residue/solvent.
13.7.1.7 Clean up the extract using the procedures in Sections 13.2 through 13.6 and 13.8. Alumina (Section 13.4) or Florisil (Section 13.8) and carbon (Section 13.5) are recommended as minimum additional cleanup steps.
13.7.1.8 Following cleanup, concentrate the extract to 10 µL as described in Section 12.7 and proceed with the analysis in Section 14.
13.7.2 Acidified silica gel (Reference 28) - Procedure alternate to the anthropogenic isolation column (Section 13.7.1) that is used for removal of lipids from the Soxhlet/SDS extraction (Section 12.4.1).
13.7.2.1 Adjust the volume of hexane in the bottle (Section 12.4.1.9.2) to approximately 200 mL.
13.7.2.2 Spike 1.0 mL of the cleanup standard (Section 7.11) into the residue/solvent.
13.7.2.3 Drop the stirring bar into the bottle, place the bottle on the stirring plate, and begin stirring.
13.7.2.4 Add 30-100 g of acid silica gel (Section 7.5.1.2) to the bottle while stirring, keeping the silica gel in motion. Stir for two to three hours.
Note:30 grams of silica gel should be adequate for most samples and will minimize contamination from this source.
13.7.2.5 After stirring, pour the extract through approximately 10 g of granular anhydrous sodium sulfate (Section 7.2.1) contained in a funnel with glass-fiber filter into a macro contration device (Section 12.6). Rinse the bottle and sodium sulfate with hexane to complete the transfer.
13.7.2.6 Concentrate the extract per Sections 12.6 through 12.7 and clean up the extract using the procedures in Sections 13.2 through 13.6 and 13.8. Alumina (Section 13.4) or Florisil (Section 13.8) and carbon (Section 13.5) are recommended as minimum additional cleanup steps.
13.7.3 Sulfuric acid and base back-extraction. Used with HCl digested extracts (Section 12.4.2).
13.7.3.1 Spike 1.0 mL of the cleanup standard (Section 7.11) into the residue/solvent (Section 12.4.2.8.2).
13.7.3.2 Add 10 mL of concentrated sulfuric acid to the bottle. Immediately cap and shake one to three times. Loosen cap in a hood to vent excess pressure. Cap and shake the bottle so that the residue/solvent is exposed to the acid for a total time of approximately 45 seconds.
13.7.3.3 Decant the hexane into a 250 mL separatory funnel making sure that no acid is transferred. Complete the quantitative transfer with several hexane rinses.
13.7.3.4 Back extract the solvent/residue with 50 mL of potassium hydroxide solution per Section 12.5.2, followed by two reagent water rinses.
13.7.3.5 Drain the extract through a filter funnel containing approximately 10 g of granular anhydrous sodium sulfate in a glass-fiber filter into a macro concentration device (Section 12.6).
13.7.3.6 Concentrate the cleaned up extract to a volume suitable for the additional cleanups given in Sections 13.2 through 13.6 and 13.8. Gel permeation chromatography (Section 13.2), alumina (Section 13.4) or Florisil (Section 13.8), and Carbopak/Celite (Section 13.5) are recommended as minimum additional cleanup steps.
13.7.3.7 Following cleanup, concentrate the extract to 10 L as described in Section 12.7 and proceed with analysis per Section 14.
13.8 Florisil Cleanup (Reference 29).
13.8.1 Pre-elute the activated Florisil column (Section 7.5.3) with 10 mL of methylene chloride followed by 10 mL of hexane:methylene chloride (98:2 v/v) and discard the solvents.
13.8.2 When the solvent is within 1 mm of the packing, apply the sample extract (in hexane) to the column. Rinse the sample container twice with 1 mL portions of hexane and apply to the column.
13.8.3 Elute the interfering compounds with 20 mL of hexane:methylene chloride (98:2) and discard the eluate.
13.8.4 Elute the CDDs/CDFs with 35 mL of methylene chloride and collect the eluate. Concentrate the eluate per Sections 12.6 through 12.7 for further cleanup or for injection into the HPLC or GC/MS.
14.0 HRGC/HRMS Analysis14.1 Establish the operating conditions given in Section 10.1.
14.2 Add 10 uL of the appropriate internal standard solution (Section 7.12) to the sample extract immediately prior to injection to minimize the possibility of loss by evaporation, adsorption, or reaction. If an extract is to be reanalyzed and evaporation has occurred, do not add more instrument internal standard solution. Rather, bring the extract back to its previous volume (e.g., 19 L) with pure nonane only (18 L if 2 L injections are used).
14.3 Inject 1.0 µL or 2.0 µL of the concentrated extract containing the internal standard solution, using on-column or splitless injection. The volume injected must be identical to the volume used for calibration (Section 10). Start the GC column initial isothermal hold upon injection. Start MS data collection after the solvent peak elutes. Stop data collection after the OCDD and OCDF have eluted. If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, stop data collection after elution of these compounds. Return the column to the initial temperature for analysis of the next extract or standard.
15.0 System and Laboratory Performance15.1 At the beginning of each 12-hour shift during which analyses are performed, GC/MS system performance and calibration are verified for all CDDs/CDFs and labeled compounds. For these tests, analysis of the CS3 calibration verification (VER) standard (Section 7.13 and Table 4) and the isomer specificity test standards (Section 7.15 and Table 5) shall be used to verify all performance criteria. Adjustment and/or recalibration (Section 10) shall be performed until all performance criteria are met. Only after all performance criteria are met may samples, blanks, IPRs, and OPRs be analyzed.
15.2 MS Resolution - A static resolving power of at least 10,000 (10% valley definition) must be demonstrated at the appropriate m/z before any analysis is performed. Static resolving power checks must be performed at the beginning and at the end of each 12-hour shift according to procedures in Section 10.1.2. Corrective actions must be implemented whenever the resolving power does not meet the requirement.
15.3 Calibration Verification.
15.3.1 Inject the VER standard using the procedure in Section 14.
15.3.2 The m/z abundance ratios for all CDDs/CDFs shall be within the limits in Table 9; otherwise, the mass spectrometer shall be adjusted until the m/z abundance ratios fall within the limits specified, and the verification test shall be repeated. If the adjustment alters the resolution of the mass spectrometer, resolution shall be verified (Section 10.1.2) prior to repeat of the verification test.
15.3.3 The peaks representing each CDD/CDF and labeled compound in the VER standard must be present with S/N of at least 10; otherwise, the mass spectrometer shall be adjusted and the verification test repeated.
15.3.4 Compute the concentration of each CDD/CDF compound by isotope dilution (Section 10.5) for those compounds that have labeled analogs (Table 1). Compute the concentration of the labeled compounds by the internal standard method (Section 10.6). These concentrations are computed based on the calibration data in Section 10.
15.3.5 For each compound, compare the concentration with the calibration verification limit in Table 6. If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, compare the concentration to the limit in Table 6a. If all compounds meet the acceptance criteria, calibration has been verified and analysis of standards and sample extracts may proceed. If, however, any compound fails its respective limit, the measurement system is not performing properly for that compound. In this event, prepare a fresh calibration standard or correct the problem causing the failure and repeat the resolution (Section 15.2) and verification (Section 15.3) tests, or recalibrate (Section 10).
15.4 Retention Times and GC Resolution.
15.4.1 Retention times.
15.4.1.1 Absolute - The absolute retention times of the 13C12-1,2,3,4-TCDD and 13C12-1,2,3,7,8,9-HxCDD GCMS internal standards in the verification test (Section 15.3) shall be within ±15 seconds of the retention times obtained during calibration (Sections 10.2.1 and 10.2.4).
15.4.1.2 Relative - The relative retention times of CDDs/CDFs and labeled compounds in the verification test (Section 15.3) shall be within the limits given in Table 2.
15.4.2 GC resolution.
15.4.2.1 Inject the isomer specificity standards (Section 7.15) on their respective columns.
15.4.2.2 The valley height between 2,3,7,8-TCDD and the other tetra-dioxin isomers at m/z 319.8965, and between 2,3,7,8-TCDF and the other tetra-furan isomers at m/z 303.9016 shall not exceed 25% on their respective columns (Figures 6 and 7).
15.4.3 If the absolute retention time of any compound is not within the limits specified or if the 2,3,7,8-isomers are not resolved, the GC is not performing properly. In this event, adjust the GC and repeat the verification test (Section 15.3) or recalibrate (Section 10), or replace the GC column and either verify calibration or recalibrate.
15.5 Ongoing Precision and Recovery.
15.5.1 Analyze the extract of the ongoing precision and recovery (OPR) aliquot (Section 11.4.2.5, 11.5.4, 11.6.2, 11.7.4, or 11.8.3.2) prior to analysis of samples from the same batch.
15.5.2 Compute the concentration of each CDD/CDF by isotope dilution for those compounds that have labeled analogs (Section 10.5). Compute the concentration of 1,2,3,7,8,9-HxCDD, OCDF, and each labeled compound by the internal standard method (Section 10.6).
15.5.3 For each CDD/CDF and labeled compound, compare the concentration to the OPR limits given in Table 6. If only 2,3,7,8-TCDD and 2,3,7,8-TCDF are to be determined, compare the concentration to the limits in Table 6a. If all compounds meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may proceed. If, however, any individual concentration falls outside of the range given, the extraction/concentration processes are not being performed properly for that compound. In this event, correct the problem, re-prepare, extract, and clean up the sample batch and repeat the ongoing precision and recovery test (Section 15.5).
15.5.4 Add results that pass the specifications in Section 15.5.3 to initial and previous ongoing data for each compound in each matrix. Update QC charts to form a graphic representation of continued laboratory performance. Develop a statement of laboratory accuracy for each CDD/CDF in each matrix type by calculating the average percent recovery (R) and the standard deviation of percent recovery (SR). Express the accuracy as a recovery interval from R−2SR to R = 2SR. For example, if R = 95% and SR = 5%, the accuracy is 85-105%.
15.6 Blank - Analyze the method blank extracted with each sample batch immediately following analysis of the OPR aliquot to demonstrate freedom from contamination and freedom from carryover from the OPR analysis. The results of the analysis of the blank must meet the specifications in Section 9.5.2 before sample analyses may proceed.
16.0 Qualitative DeterminationA CDD, CDF, or labeled compound is identified in a standard, blank, or sample when all of the criteria in Sections 16.1 through 16.4 are met.
16.1 The signals for the two exact m/z's in Table 8 must be present and must maximize within the same two seconds.
16.2 The signal-to-noise ratio (S/N) for the GC peak at each exact m/z must be greater than or equal to 2.5 for each CDD or CDF detected in a sample extract, and greater than or equal to 10 for all CDDs/CDFs in the calibration standard (Sections 10.2.3 and 15.3.3).
16.3 The ratio of the integrated areas of the two exact m/z's specified in Table 8 must be within the limit in Table 9, or within ±10% of the ratio in the midpoint (CS3) calibration or calibration verification (VER), whichever is most recent.
16.4 The relative retention time of the peak for a 2,3,7,8-substituted CDD or CDF must be within the limit in Table 2. The retention time of peaks representing non-2,3,7,8-substituted CDDs/CDFs must be within the retention time windows established in Section 10.3.
16.5 Confirmatory Analysis - Isomer specificity for 2,3,7,8-TCDF cannot be achieved on the DB-5 column. Therefore, any sample in which 2,3,7,8-TCDF is identified by analysis on a DB-5 column must have a confirmatory analysis performed on a DB-225, SP-2330, or equivalent GC column. The operating conditions in Section 10.1.1 may be adjusted to optimize the analysis on the second GC column, but the GC/MS must meet the mass resolution and calibration specifications in Section 10.
16.6 If the criteria for identification in Sections 16.1 through 16.5 are not met, the CDD or CDF has not been identified and the results may not be reported for regulatory compliance purposes. If interferences preclude identification, a new aliquot of sample must be extracted, further cleaned up, and analyzed.
17.0 Quantitative Determination
17.1 Isotope Dilution Quantitation - By adding a known amount of a labeled compound to every sample prior to extraction, correction for recovery of the CDD/CDF can be made because the CDD/CDF and its labeled analog exhibit similar effects upon extraction, concentration, and gas chromatography. Relative response (RR) values are used in conjunction with the initial calibration data described in Section 10.5 to determine concentrations directly, so long as labeled compound spiking levels are constant, using the following equation:
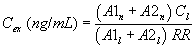 where:
Cex = The concentration of the CDD/CDF in the extract, and the
other terms are as defined in Section 10.5.2.
where:
Cex = The concentration of the CDD/CDF in the extract, and the
other terms are as defined in Section 10.5.2.
17.1.1 Because of a potential interference, the labeled analog of OCDF is not added to the sample. Therefore, OCDF is quantitated against labeled OCDD. As a result, the concentration of OCDF is corrected for the recovery of the labeled OCDD. In instances where OCDD and OCDF behave differently during sample extraction, concentration, and cleanup procedures, this may decrease the accuracy of the OCDF results. However, given the low toxicity of this compound relative to the other dioxins and furans, the potential decrease in accuracy is not considered significant.
17.1.2 Because 13C12-1,2,3,7,8,9-HxCDD is used as an instrument internal standard (i.e., not added before extraction of the sample), it cannot be used to quantitate the 1,2,3,7,8,9-HxCDD by strict isotope dilution procedures. Therefore, 1,2,3,7,8,9-HxCDD is quantitated using the averaged response of the labeled analogs of the other two 2,3,7,8-substituted HxCDD's: 1,2,3,4,7,8-HxCDD and 1,2,3,6,7,8-HxCDD. As a result, the concentration of 1,2,3,7,8,9-HxCDD is corrected for the average recovery of the other two HxCDD's.
17.1.3 Any peaks representing non-2,3,7,8-substituted CDDs/CDFs are quantitated using an average of the response factors from all of the labeled 2,3,7,8-isomers at the same level of chlorination.
17.2 Internal Standard Quantitation and Labeled Compound Recovery.
17.2.1 Compute the concentrations of 1,2,3,7,8,9-HxCDD, OCDF, the 13C-labeled analogs and the 37C-labeled cleanup standard in the extract using the response factors determined from the initial calibration data (Section 10.6) and the following equation:
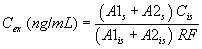 where:
Cex = The concentration of the CDD/CDF in the extract, and the
other terms are as defined in Section 10.6.1. Note:
where:
Cex = The concentration of the CDD/CDF in the extract, and the
other terms are as defined in Section 10.6.1. Note:
There is only one m/z for the 37Cl-labeled standard.
17.2.2 Using the concentration in the extract determined above, compute the percent recovery of the 13C-labeled compounds and the 37C-labeled cleanup standard using the following equation:

17.3 The concentration of a CDD/CDF in the solid phase of the sample is computed using the concentration of the compound in the extract and the weight of the solids (Section 11.5.1), as follows:
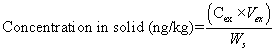 where:
Cex = The concentration of the compound in the extract. Vex = The
extract volume in mL. Ws = The sample weight (dry weight) in kg.
where:
Cex = The concentration of the compound in the extract. Vex = The
extract volume in mL. Ws = The sample weight (dry weight) in kg.
17.4 The concentration of a CDD/CDF in the aqueous phase of the sample is computed using the concentration of the compound in the extract and the volume of water extracted (Section 11.4 or 11.5), as follows:
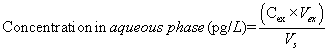 where:
Cex = The concentration of the compound in the extract. Vex = The
extract volume in mL. Vs = The sample volume in liters.
where:
Cex = The concentration of the compound in the extract. Vex = The
extract volume in mL. Vs = The sample volume in liters.
17.5 If the SICP area at either quantitation m/z for any compound exceeds the calibration range of the system, a smaller sample aliquot is extracted.
17.5.1 For aqueous samples containing 1% solids or less, dilute 100 mL, 10 mL, etc., of sample to 1 L with reagent water and re-prepare, extract, clean up, and analyze per Sections 11 through 14.
17.5.2 For samples containing greater than 1% solids, extract an amount of sample equal to 1/10, 1/100, etc., of the amount used in Section 11.5.1. Re-prepare, extract, clean up, and analyze per Sections 11 through 14.
17.5.3 If a smaller sample size will not be representative of the entire sample, dilute the sample extract by a factor of 10, adjust the concentration of the instrument internal standard to 100 pg/µL in the extract, and analyze an aliquot of this diluted extract by the internal standard method.
17.6 Results are reported to three significant figures for the CDDs/CDFs and labeled compounds found in all standards, blanks, and samples.
17.6.1 Reporting units and levels.
17.6.1.1 Aqueous samples - Report results in pg/L (parts-per-quadrillion).
17.6.1.2 Samples containing greater than 1% solids (soils, sediments, filter cake, compost) - Report results in ng/kg based on the dry weight of the sample. Report the percent solids so that the result may be corrected.
17.6.1.3 Tissues - Report results in ng/kg of wet tissue, not on the basis of the lipid content of the sample. Report the percent lipid content, so that the data user can calculate the concentration on a lipid basis if desired.
17.6.1.4 Reporting level.
17.6.1.4.1 Standards (VER, IPR, OPR) and samples - Report results at or above the minimum level (Table 2). Report results below the minimum level as not detected or as required by the regulatory authority.
17.6.1.4.2 Blanks - Report results above one-third the ML.
17.6.2 Results for CDDs/CDFs in samples that have been diluted are reported at the least dilute level at which the areas at the quantitation m/z's are within the calibration range (Section 17.5).
17.6.3 For CDDs/CDFs having a labeled analog, results are reported at the least dilute level at which the area at the quantitation m/z is within the calibration range (Section 17.5) and the labeled compound recovery is within the normal range for the method (Section 9.3 and Tables 6, 6a, 7, and 7a).
17.6.4 Additionally, if requested, the total concentration of all isomers in an individual level of chlorination (i.e., total TCDD, total TCDF, total Paced, etc.) may be reported by summing the concentrations of all isomers identified in that level of chlorination, including both 2,3,7,8-substituted and non-2,3,7,8-substituted isomers.
18.0 Analysis of Complex Samples18.1 Some samples may contain high levels (>10 ng/L; >1000 ng/kg) of the compounds of interest, interfering compounds, and/or polymeric materials. Some extracts will not concentrate to 10 µL (Section 12.7); others may overload the GC column and/or mass spectrometer.
18.2 Analyze a smaller aliquot of the sample (Section 17.5) when the extract will not concentrate to 10 µL after all cleanup procedures have been exhausted.
18.3 Chlorodiphenyl Ethers - If chromatographic peaks are detected at the retention time of any CDDs/CDFs in any of the m/z channels being monitored for the chlorodiphenyl ethers (Table 8), cleanup procedures must be employed until these interferences are removed. Alumina (Section 13.4) and Florisil (Section 13.8) are recommended for removal of chlorodiphenyl ethers.
18.4 Recovery of Labeled Compounds - In most samples, recoveries of the labeled compounds will be similar to those from reagent water or from the alternate matrix (Section 7.6).
18.4.1 If the recovery of any of the labeled compounds is outside of the normal range (Table 7), a diluted sample shall be analyzed (Section 17.5).
18.4.2 If the recovery of any of the labeled compounds in the diluted sample is outside of normal range, the calibration verification standard (Section 7.13) shall be analyzed and calibration verified (Section 15.3).
18.4.3 If the calibration cannot be verified, a new calibration must be performed and the original sample extract reanalyzed.
18.4.4 If the calibration is verified and the diluted sample does not meet the limits for labeled compound recovery, the method does not apply to the sample being analyzed and the result may not be reported for regulatory compliance purposes. In this case, alternate extraction and cleanup procedures in this method must be employed to resolve the interference. If all cleanup procedures in this method have been employed and labeled compound recovery remains outside of the normal range, extraction and/or cleanup procedures that are beyond this scope of this method will be required to analyze these samples.
19.0 Pollution Prevention19.1 The solvents used in this method pose little threat to the environment when managed properly. The solvent evaporation techniques used in this method are amenable to solvent recovery, and it is recommended that the laboratory recover solvents wherever feasible.
19.2 Standards should be prepared in volumes consistent with laboratory use to minimize disposal of standards.
20.0 Waste Management20.1 It is the laboratory's responsibility to comply with all federal, state, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions, and to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance is also required with any sewage discharge permits and regulations.
20.2 Samples containing HCl to pH <2 are hazardous and must be neutralized before being poured down a drain or must be handled as hazardous waste.
20.3 The CDDs/CDFs decompose above 800 °C. Low-level waste such as absorbent paper, tissues, animal remains, and plastic gloves may be burned in an appropriate incinerator. Gross quantities (milligrams) should be packaged securely and disposed of through commercial or governmental channels that are capable of handling extremely toxic wastes.
20.4 Liquid or soluble waste should be dissolved in methanol or ethanol and irradiated with ultraviolet light with a wavelength shorter than 290 nm for several days. Use F40 BL or equivalent lamps. Analyze liquid wastes, and dispose of the solutions when the CDDs/CDFs can no longer be detected.
20.5 For further information on waste management, consult “The Waste Management Manual for Laboratory Personnel” and “Less is Better - Laboratory Chemical Management for Waste Reduction,” available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street N.W., Washington, D.C. 20036.
21.0 Method PerformanceMethod performance was validated and performance specifications were developed using data from EPA's international interlaboratory validation study (References 30-31) and the EPA/paper industry Long-Term Variability Study of discharges from the pulp and paper industry (58 FR 66078).
22.0 References1. Tondeur, Yves. “Method 8290: Analytical Procedures and Quality Assurance for Multimedia Analysis of Polychlorinated Dibenzo-p-dioxins and Dibenzofurans by High Resolution Gas Chromatography/High Resolution Mass Spectrometry,” USEPA EMSL, Las Vegas, Nevada, June 1987.
2. “Measurement of 2,3,7,8-Tetrachlorinated Dibenzo-p-dioxin (TCDD) and 2,3,7,8-Tetrachlorinated Dibenzofuran (TCDF) in Pulp, Sludges, Process Samples and Wastewaters from Pulp and Paper Mills,” Wright State University, Dayton, OH 45435, June 1988.
3. “NCASI Procedures for the Preparation and Isomer Specific Analysis of Pulp and Paper Industry Samples for 2,3,7,8-TCDD and 2,3,7,8-TCDF,” National Council of the Paper Industry for Air and Stream Improvement Inc., 260 Madison Avenue, New York, NY 10016, Technical Bulletin No. 551, Pre-Release Copy, July 1988.
4. “Analytical Procedures and Quality Assurance Plan for the Determination of PCDD/PCDF in Fish,” USEPA, Environmental Research Laboratory, 6201 Congdon Boulevard, Duluth, MN 55804, April 1988.
5. Tondeur, Yves. “Proposed GC/MS Methodology for the Analysis of PCDDs and PCDFs in Special Analytical Services Samples,” Triangle Laboratories, Inc., 801-10 Capitola Dr, Research Triangle Park, NC 27713, January 1988; updated by personal communication September 1988.
6. Lamparski, L.L. and Nestrick, T.J. “Determination of Tetra-, Hexa-, Hepta-, and Octachlorodibenzo-p-dioxin Isomers in Particulate Samples at Parts per Trillion Levels,” Analytical Chemistry, 52: 2045-2054, 1980.
7. Lamparski, L.L. and Nestrick, T.J. “Novel Extraction Device for the Determination of Chlorinated Dibenzo-p-dioxins (PCDDs) and Dibenzofurans (PCDFs) in Matrices Containing Water,” Chemosphere, 19:27-31, 1989.
8. Patterson, D.G., et. al. “Control of Interferences in the Analysis of Human Adipose Tissue for 2,3,7,8-Tetrachlorodibenzo-p-dioxin,” Environmental Toxicological Chemistry, 5:355-360, 1986.
9. Stanley, John S. and Sack, Thomas M. “Protocol for the Analysis of 2,3,7,8-Tetrachlorodibenzo-p-dioxin by High Resolution Gas Chromatography/High Resolution Mass Spectrometry,” USEPA EMSL, Las Vegas, Nevada 89114, EPA 600/4-86-004, January 1986.
10. “Working with Carcinogens,” Department of Health, Education, & Welfare, Public Health Service, Centers for Disease Control, NIOSH, Publication 77-206, August 1977, NTIS PB-277256.
11. “OSHA Safety and Health Standards, General Industry,” OSHA 2206, 29 CFR 1910.
12. “Safety in Academic Chemistry Laboratories,” ACS Committee on Chemical Safety, 1979.
13. “Standard Methods for the Examination of Water and Wastewater,” 18th edition and later revisions, American Public Health Association, 1015 15th St, N.W., Washington, DC 20005, 1-35: Section 1090 (Safety), 1992.
14. “Method 613 - 2,3,7,8-Tetrachlorodibenzo-p-dioxin,” 40 CFR 136 (49 FR 43234), October 26, 1984, Section 4.1.
15. Provost, L.P. and Elder, R.S. “Interpretation of Percent Recovery Data,” American Laboratory, 15: 56-83, 1983.
16. “Standard Practice for Sampling Water,” ASTM Annual Book of Standards, ASTM, 1916 Race Street, Philadelphia, PA 19103-1187, 1980.
17. “Methods 330.4 and 330.5 for Total Residual Chlorine,” USEPA, EMSL, Cincinnati, OH 45268, EPA 600/4-79-020, March 1979.
18. “Handbook of Analytical Quality Control in Water and Wastewater Laboratories,” USEPA EMSL, Cincinnati, OH 45268, EPA-600/4-79-019, March 1979.
19. Williams, Rick. Letter to Bill Telliard, June 4, 1993, available from the EPA Sample Control Center operated by DynCorp Viar, Inc., 300 N Lee St, Alexandria, VA 22314, 703-519-1140.
20. Barkowski, Sarah. Fax to Sue Price, August 6, 1992, available from the EPA Sample Control Center operated by DynCorp Viar, Inc., 300 N Lee St, Alexandria VA 22314, 703-519-1140.
21. “Analysis of Multi-media, Multi-concentration Samples for Dioxins and Furans, PCDD/PCDF Analyses Data Package”, Narrative for Episode 4419, MRI Project No. 3091-A, op.cit. February 12, 1993, Available from the EPA Sample Control Center operated by DynCorp Viar Inc, 300 N Lee St, Alexandria, VA 22314 (703-519-1140).
22. “Analytical Procedures and Quality Assurance Plan for the Determination of PCDD/PCDF in Fish”, U.S. Environmental Protection Agency, Environmental Research Laboratory, Duluth, MN 55804, EPA/600/3-90/022, March 1990.
23. Afghan, B.K., Carron, J., Goulden, P.D., Lawrence, J., Leger, D., Onuska, F., Sherry, J., and Wilkenson, R.J., “Recent Advances in Ultratrace Analysis of Dioxins and Related Halogenated Hydrocarbons”, Can J. Chem., 65: 1086-1097, 1987.
24. Sherry, J.P. and Tse, H. “A Procedure for the Determination of Polychlorinated Dibenzo-p-dioxins in Fish”, Chemosphere, 20: 865-872, 1990.
25. “Preliminary Fish Tissue Study”, Results of Episode 4419, available from the EPA Sample Control Center operated by DynCorp Viar, Inc., 300 N Lee St, Alexandria, VA 22314, 703-519-1140.
26. Nestrick, Terry L. DOW Chemical Co., personal communication with D.R. Rushneck, April 8, 1993. Details available from the U.S. Environmental Protection Agency Sample Control Center operated by DynCorp Viar Inc, 300 N Lee St, Alexandria, VA 22314, 703-519-1140.
27. Barnstadt, Michael. “Big Fish Column”, Triangle Laboratories of RTP, Inc., SOP 129-90, 27 March 27, 1992.
28. “Determination of Polychlorinated Dibenzo-p-Dioxins (PCDD) and Dibenzofurans (PCDF) in Environmental Samples Using EPA Method 1613”, Chemical Sciences Department, Midwest Research Institute, 425 Volker Boulevard, Kansas City, MO 44110-2299, Standard Operating Procedure No. CS-153, January 15, 1992.
29. Ryan, John J. Raymonde Lizotte and William H. Newsome, J. Chromatog. 303 (1984) 351-360.
30. Telliard, William A., McCarty, Harry B., and Riddick, Lynn S. “Results of the Interlaboratory Validation Study of USEPA Method 1613 for the Analysis of Tetra-through Octachlorinated Dioxins and Furans by Isotope Dilution GC/MS,” Chemosphere, 27, 41-46 (1993).
31. “Results of the International Interlaboratory Validation Study of USEPA Method 1613”, October 1994, available from the EPA Sample Control Center operated by DynCorp Viar, Inc., 300 N Lee St, Alexandria, VA 22314, 703-519-1140.
23.0 Tables and FiguresTable 1 - Chlorinated Dibenzo-p-Dioxins and Furans Determined by Isotope Dilution and Internal Standard High Resolution Gas Chromatography (HRGC)/High Resolution Mass Spectrometry (HRMS)
| CDDs/CDFs 1 | CAS registry | Labeled analog | CAS registry |
|---|---|---|---|
| 2,3,7,8-TCDD | 1746-01-6 |
13C12-2,3,7,8-TCDD 37Cl4-2,3,7,8-TCDD |
76523-40-5 85508-50-5 |
| Total TCDD | 41903-57-5 | ||
| 2,3,7,8-TCDF | 51207-31-9 | 13C12-2,3,7,8-TCDF | 89059-46-1 |
| Total-TCDF | 55722-27-5 | ||
| 1,2,3,7,8-PeCDD | 40321-76-4 | 13C12-1,2,3,7,8-PeCDD | 109719-79-1 |
| Total-PeCDD | 36088-22-9 | ||
| 1,2,3,7,8-PeCDF | 57117-41-6 | 13C12-1,2,3,7,8-PeCDF | 109719-77-9 |
| 2,3,4,7,8-PeCDF | 57117-31-4 | 13C12-2,3,4,7,8-PeCDF | 116843-02-8 |
| Total-PeCDF | 30402-15-4 | ||
| 1,2,3,4,7,8-HxCDD | 39227-28-6 | 13C12-1,2,3,4,7,8-HxCDD | 109719-80-4 |
| 1,2,3,6,7,8-HxCDD | 57653-85-7 | 13C12-1,2,3,6,7,8-HxCDD | 109719-81-5 |
| 1,2,3,7,8,9-HxCDD | 19408-74-3 | 13C12-1,2,3,7,8,9-HxCDD | 109719-82-6 |
| Total-HxCDD | 34465-46-8 | ||
| 1,2,3,4,7,8-HxCDF | 70648-26-9 | 13C12-1,2,3,4,7,8-HxCDF | 114423-98-2 |
| 1,2,3,6,7,8-HxCDF | 57117-44-9 | 13C12-1,2,3,6,7,8-HxCDF | 116843-03-9 |
| 1,2,3,7,8,9-HxCDF | 72918-21-9 | 13C12-1,2,3,7,8,9-HxCDF | 116843-04-0 |
| 2,3,4,6,7,8-HxCDF | 60851-34-5 | 13C12-2,3,4,6,7,8-HxCDF | 116843-05-1 |
| Total-HxCDF | 55684-94-1 | ||
| 1,2,3,4,6,7,8-HpCDD | 35822-46-9 | 13C12-1,2,3,4,6,7,8-HpCDD | 109719-83-7 |
| Total-HpCDD | 37871-00-4 | ||
| 1,2,3,4,6,7,8-HpCDF | 67562-39-4 | 13C12-1,2,3,4,6,7,8-HpCDF | 109719-84-8 |
| 1,2,3,4,7,8,9-HpCDF | 55673-89-7 | 13C12-1,2,3,4,7,8,9-HpCDF | 109719-94-0 |
| Total-HpCDF | 38998-75-3 | ||
| OCDD | 3268-87-9 | 13C12-OCDD | 114423-97-1 |
| OCDF | 39001-02-0 | Not used |
1 Chlorinated dibenzo-p-dioxins and chlorinated dibenzofurans.
TCDD = Tetrachlorodibenzo-p-dioxin.
TCDF = Tetrachlorodibenzofuran.
PeCDD = Pentachlorodibenzo-p-dioxin.
PeCDF = Pentachlorodibenzofuran.
HxCDD = Hexachlorodibenzo-p-dioxin.
HxCDF = Hexachlorodibenzofuran.
HpCDD = Heptachlorodibenzo-p-dioxin.
HpCDF = Heptachlorodibenzofuran.
OCDD = Octachlorodibenzo-p-dioxin.
OCDF = Octachlorodibenzofuran.
Table 2 - Retention Time References, Quantitation References, Relative Retention Times, and Minimum Levels for CDDS and DCFS
| CDD/CDF | Retention time
and quantitation reference |
Relative retention time | Minimum level 1 | ||
|---|---|---|---|---|---|
| Water (pg/L; ppq) | Solid (ng/kg; ppt) | Extract (pg/µL; ppb) | |||
| Compounds using 13 C12-1,2,3,4-TCDD as the Injection Internal Standard | |||||
| 2,3,7,8-TCDF | 13 C12-2,3,7,8-TCDF | 0.999-1.003 | 10 | 1 | 0.5 |
| 2,3,7,8-TCDD | 13 C12-2,3,7,8-TCDD | 0.999-1.002 | 10 | 1 | 0.5 |
| 1,2,3,7,8-Pe | 13 C12-1,2,3,7,8-PeCDF | 0.999-1.002 | 50 | 5 | 2.5 |
| 2,3,4,7,8-PeCDF | 13 C12-2,3,4,7,8-PeCDF | 0.999-1.002 | 50 | 5 | 2.5 |
| 1,2,3,7,8-PeCDD | 13 C12-1,2,3,7,8-PeCDD | 0.999-1.002 | 50 | 5 | 2.5 |
| 13 C12-2,3,7,8-TCDF | 13 C12-1,2,3,4-TCDD | 0.923-1.103 | |||
| 13 C12-2,3,7,8-TCDD | 13 C12-1,2,3,4-TCDD | 0.976-1.043 | |||
| 13 C12-2,3,7,8-TCDD | 13 C12-1,2,3,4-TCDD | 0.989-1.052 | |||
| 13 C12-1,2,3,7,8-PeCDF | 13 C12-1,2,3,4-TCDD | 1.000-1.425 | |||
| 13 C12-2,3,4,7,8-PeCDF | 13 C12-1,2,3,4-TCDD | 1.001-1.526 | |||
| 13 C12-1,2,3,7,8-PeCDF | 13 C12-1,2,3,4-TCDD | 1.000-1.567 | |||
| Compounds using 13 C12-1,2,3,7,8,9-HxCDD as the Injection Internal Standard | |||||
| 1,2,3,4,7,8-HxCDF | 13 C12-1,2,3,4,7,8-HxCDF | 0.999-1.001 | 50 | 5 | 2.5 |
| 1,2,3,6,7,8-HxCDF | 13 C12-1,2,3,6,7,8-HxCDF | 0.997-1.005 | 50 | 5 | 2.5 |
| 1,2,3,7,8,9-HxCDF | 13 C12-1,2,3,7,8,9-HxCDF | 0.999-1.001 | 50 | 5 | 2.5 |
| 2,3,4,6,7,8-HxCDF | 13 C12-2,3,4,6,7,8-HxCDF | 0.999-1.001 | 50 | 5 | 2.5 |
| 1,2,3,4,7,8-HxCDD | 13 C12-1,2,3,4,7,8-HxCDD | 0.999-1.001 | 50 | 5 | 2.5 |
| 1,2,3,6,7,8-HxCDD | 13 C12-1,2,3,6,7,8-HxCDD | 0.998-1.004 | 50 | 5 | 2.5 |
| 1,2,3,7,8,9-HxCDD | ( 2) | 1.000-1.019 | 50 | 5 | 2.5 |
| 1,2,3,4,6,7,8-HpCDF | 13 C12-1,2,3,4,6,7,8-HpCDF | 0.999-1.001 | 50 | 5 | 2.5 |
| 1,2,3,4,7,8,9-HpCDF | 13 C12-1,2,3,4,7,8,9-HpCDF | 0.999-1.001 | 50 | 5 | 2.5 |
| 1,2,3,4,6,7,8-HpCDD | 13 C12-1,2,3,4,6,7,8-HpCDD | 0.999-1.001 | 50 | 5 | 2.5 |
| OCDF | 13 C12-OCDD | 0.999-1.001 | 100 | 10 | 5.0 |
| OCDD | 13 C12-OCDD | 0.999-1.001 | 100 | 10 | 5.0 |
| 1,2,3,4,6,7,8,-HxCDF | 13 C12-1,2,3,7,8,9-HpCDD | 0.949-0.975 | |||
| 13 C121,2,3,7,8,9-HxCDF | 13 C12-1,2,3,7,8,9-HpCDD | 0.977-1.047 | |||
| 13 C122,3,4,6,7,8,-HxCDF | 13 C12-1,2,3,7,8,9-HpCDD | 0.959-1.021 | |||
| 13 C121,2,3,4,7,8,-HxCDF | 13 C12-1,2,3,7,8,9-HpCDD | 0.977-1.000 | |||
| 13 C121,2,3,6,7,8,-HxCDF | 13 C12-1,2,3,7,8,9-HpCDD | 0.981-1.003 | |||
| 13 C121,2,3,4,6,7,8-HxCDF | 13 C12-1,2,3,7,8,9-HpCDD | 1.043-1.085 | |||
| 13 C121,2,3,4,7,8,9-HxCDF | 13 C12-1,2,3,7,8,9-HpCDD | 1.057-1.151 | |||
| 13 C121,2,3,4,6,7,8-HxCDF | 13 C12-1,2,3,7,8,9-HpCDD | 1.086-1.110 | |||
| 13 C12OCDD | 13 C12-1,2,3,7,8,9-HpCDD | 1.032-1.311 | |||
1 The Minimum Level (ML) for each analyte is defined as the level at which the entire analytical system must give a recognizable signal and acceptable calibration point. It is equivalent to the concentration of the lowest calibration standard, assuming that all method-specified sample weights, volumes, and cleanup procedures have been employed.
2 The retention time reference for 1,2,3,7,8,9-HxCDD is 13C12-1,2,3,6,7,8-HxCDD, and 1,2,3,7,8,9-HxCDD is quantified using the averaged responses for 13C12-1,2,3,4,7,8-HxCDD and 13C12-1,2,3,6,7,8-HxCDD.
Table 3 - Concentration of Stock and Spiking Solutions Containing CDDS/CDFS and Labeled Compounds
| CDD/CDF | Labeled compound stock
solution 1 (ng/mL) |
Labeled compound spiking
solution 2 (ng/mL) |
PAR stock solution 3 (ng/mL) | PAR spiking solution
4 (ng/mL) |
|---|---|---|---|---|
| 2,3,7,8-TCDD | 40 | 0.8 | ||
| 2,3,7,8-TCDF | 40 | 0.8 | ||
| 1,2,3,7,8-PeCDD | 200 | 4 | ||
| 1,2,3,7,8-PeCDF | 200 | 4 | ||
| 2,3,4,7,8-PeCDF | 200 | 4 | ||
| 1,2,3,4,7,8-HxCDD | 200 | 4 | ||
| 1,2,3,6,7,8-HxCDD | 200 | 4 | ||
| 1,2,3,7,8,9-HxCDD | 200 | 4 | ||
| 1,2,3,4,7,8-HxCDF | 200 | 4 | ||
| 1,2,3,6,7,8-HxCDF | 200 | 4 | ||
| 1,2,3,7,8,9-HxCDF | 200 | 4 | ||
| 2,3,4,6,7,8-HxCDF | 200 | 4 | ||
| 1,2,3,4,6,7,8-HpCDD | 200 | 4 | ||
| 1,2,3,4,6,7,8-HpCDF | 200 | 4 | ||
| 1,2,3,4,7,8,9-HpCDF | 200 | 4 | ||
| OCDD | 400 | 8 | ||
| OCDF | 400 | 8 | ||
| 13C12-2,3,7,8-TCDD | 100 | 2 | ||
| 13C12-2,3,7,8-TCDF | 100 | 2 | ||
| 13C12-1,2,3,7,8-PeCDD | 100 | 2 | ||
| 13C12-1,2,3,7,8-PeCDF | 100 | 2 | ||
| 13C12-2,3,4,7,8-PeCDF | 100 | 2 | ||
| 13C12-1,2,3,4,7,8-HxCDD | 100 | 2 | ||
| 13C12-1,2,3,6,7,8-HxCDD | 100 | 2 | ||
| 13C12-1,2,3,4,7,8-HxCDF | 100 | 2 | ||
| 13C12-1,2,3,6,7,8-HxCDF | 100 | 2 | ||
| 13C12-1,2,3,7,8,9-HxCDF | 100 | 2 | ||
| 13C12-2,3,4,6,7,8-HxCDF | 100 | 2 | ||
| 13C12-1,2,3,4,6,7,8-HpCDD | 100 | 2 | ||
| 13C12-1,2,3,4,6,7,8-HpCDF | 100 | 2 | ||
| 13C12-1,2,3,4,7,8,9-HpCDF | 100 | 2 | ||
| 13C12-OCDD | 200 | 4 | ||
| Cleanup Standard 5 | ||||
| 37Cl4-2,3,7,8-TCDD | 0.8 | |||
| Internal Standards 6 | ||||
| 13C12-1,2,3,4-TCDD | 200 | |||
| 13C12-1,2,3,7,8,9-HxCDD | 200 |
1 Section 7.10 - prepared in nonane and diluted to prepare spiking solution.
2 Section 7.10.3 - prepared in acetone from stock solution daily.
3 Section 7.9 - prepared in nonane and diluted to prepare spiking solution.
4 Section 7.14 - prepared in acetone from stock solution daily.
5 Section 7.11 - prepared in nonane and added to extract prior to cleanup.
6 Section 7.12 - prepared in nonane and added to the concentrated extract immediately prior to injection into the GC (Section 14.2).
Table 4 - Concentration of CDDS/CDFS in Calibration and Calibration Verification Solutions 1 (Section 15.3)
| CDD/CDF | CS2 (ng/mL) |
CS3 (ng/mL) |
CS4 (ng/mL) |
CS5 (ng/mL) |
|
|---|---|---|---|---|---|
| 2,3,7,8-TCDD | 0.5 | 2 | 10 | 40 | 200 |
| 2,3,7,8-TCDF | 0.5 | 2 | 10 | 40 | 200 |
| 1,2,3,7,8-PeCDD | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,7,8-PeCDF | 2.5 | 10 | 50 | 200 | 1000 |
| 2,3,4,7,8-PeCDF | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,4,7,8-HxCDD | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,6,7,8-HxCDD | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,7,8,9-HxCDD | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,4,7,8-HxCDF | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,6,7,8-HxCDF | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,7,8,9-HxCDF | 2.5 | 10 | 50 | 200 | 1000 |
| 2,3,4,6,7,8-HxCDF | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,4,6,7,8-HpCDD | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,4,6,7,8-HpCDF | 2.5 | 10 | 50 | 200 | 1000 |
| 1,2,3,4,7,8,9-HpCDF | 2.5 | 10 | 50 | 200 | 1000 |
| OCDD | 5.0 | 20 | 100 | 400 | 2000 |
| OCDF | 5.0 | 20 | 100 | 400 | 2000 |
| 13 C12-2,3,7,8-TCDD | 100 | 100 | 100 | 100 | 100 |
| 13 C12-2,3,7,8-TCDF | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,7,8-PeCDD | 100 | 100 | 100 | 100 | 100 |
| 13 C12-PeCDF | 100 | 100 | 100 | 100 | 100 |
| 13 C12-2,3,4,7,8-PeCDF | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,4,7,8-HxCDD | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,6,7,8-HxCDD | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,4,7,8-HxCDF | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,6,7,8-HxCDF | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,7,8,9-HxCDF | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,4,6,7,8-HpCDD | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,4,6,7,8-HpCDF | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,4,7,8,9-Hp CDF | 100 | 100 | 100 | 100 | 100 |
| 13 C12-OCDD | 200 | 200 | 200 | 200 | 200 |
| Cleanup Standard: | |||||
| 37 C14-2,3,7,8-TCDD | 0.5 | 2 | 10 | 40 | 200 |
| Internal Standards: | |||||
| 13 C12-1,2,3,4-TCDD | 100 | 100 | 100 | 100 | 100 |
| 13 C12-1,2,3,7,8,9-HxCDD | 100 | 100 | 100 | 100 | 100 |
Table 5 - GC Retention Time Window Defining Solution and Isomer Specificity Test Standard (Section 7.15)
| DB-5 column GC retention-time window defining solution | ||
|---|---|---|
| CDD/CDF | First eluted | Last eluted |
| TCDF | 1,3,6,8- | 1,2,8,9- |
| TCDD | 1,3,6,8- | 1,2,8,9- |
| PeCDF | 1,3,4,6,8- | 1,2,3,8,9- |
| PeCDD | 1,2,4,7,9- | 1,2,3,8,9- |
| HxCDF | 1,2,3,4,6,8- | 1,2,3,4,8,9- |
| HxCDD | 1,2,4,6,7,9- | 1,2,3,4,6,7- |
| HpCDF | 1,2,3,4,6,7,8- | 1,2,3,4,7,8,9- |
| HpCDD | 1,2,3,4,6,7,9- | 1,2,3,4,6,7,8- |
| DB-5 Column TCDD Specificity Test Standard |
| 1,2,3,7 = 1,2,3,8-TCDD |
| 2,3,7,8-TCDD |
| 1,2,3,9-TCDD |
| DB-225 Column TCDF Isomer Specificity Test Standard |
| 2,3,4,7-TCDF |
| 2,3,7,8-TCDF |
| 1,2,3,9-TCDF |
Table 6 - Acceptance Criteria for Performance Tests When All CDDS/CDFS Are Tested 1
| CDD/CDF | Test conc. (ng/mL) | IPR 2 3 | OPR (ng/mL) |
VER (ng/mL) |
|
|---|---|---|---|---|---|
| s (ng/mL) |
X (ng/mL) |
||||
| 2,3,7,8-TCDD | 10 | 2.8 | 8.3-12.9 | 6.7-15.8 | 7.8-12.9 |
| 2,3,7,8-TCDF | 10 | 2.0 | 8.7-13.7 | 7.5-15.8 | 8.4-12.0 |
| 1,2,3,7,8-PeCDD | 50 | 7.5 | 38-66 | 35-71 | 39-65 |
| 1,2,3,7,8-PeCDF | 50 | 7.5 | 43-62 | 40-67 | 41-60 |
| 2,3,4,7,8-PeCDF | 50 | 8.6 | 36-75 | 34-80 | 41-61 |
| 1,2,3,4,7,8-HxCDD | 50 | 9.4 | 39-76 | 35-82 | 39-64 |
| 1,2,3,6,7,8-HxCDD | 50 | 7.7 | 42-62 | 38-67 | 39-64 |
| 1,2,3,7,8,9-HxCDD | 50 | 11.1 | 37-71 | 32-81 | 41-61 |
| 1,2,3,4,7,8-HxCDF | 50 | 8.7 | 41-59 | 36-67 | 45-56 |
| 1,2,3,6,7,8-HxCDF | 50 | 6.7 | 46-60 | 42-65 | 44-57 |
| 1,2,3,7,8,9-HxCDF | 50 | 6.4 | 42-61 | 39-65 | 45-56 |
| 2,3,4,6,7,8-HxCDF | 50 | 7.4 | 37-74 | 35-78 | 44-57 |
| 1,2,3,4,6,7,8-HpCDD | 50 | 7.7 | 38-65 | 35-70 | 43-58 |
| 1,2,3,4,6,7,8-HpCDF | 50 | 6.3 | 45-56 | 41-61 | 45-55 |
| 1,2,3,4,7,8,9-HpCDF | 50 | 8.1 | 43-63 | 39-69 | 43-58 |
| OCDD | 100 | 19 | 89-127 | 78-144 | 79-126 |
| OCDF | 100 | 27 | 74-146 | 63-170 | 63-159 |
| 13C12-2,3,7,8-TCDD | 100 | 37 | 28-134 | 20-175 | 82-121 |
| 13C12-2,3,7,8-TCDF | 100 | 35 | 31-113 | 22-152 | 71-140 |
| 13C12-1,2,3,7,8-PeCDD | 100 | 39 | 27-184 | 21-227 | 62-160 |
| 13C12-1,2,3,7,8-PeCDF | 100 | 34 | 27-156 | 21-192 | 76-130 |
| 13C12-2,3,4,7,8-PeCDF | 100 | 38 | 16-279 | 13-328 | 77-130 |
| 13C12-1,2,3,4,7,8-HxCDD | 100 | 41 | 29-147 | 21-193 | 85-117 |
| 13C12-1,2,3,6,7,8-HxCDD | 100 | 38 | 34-122 | 25-163 | 85-118 |
| 13C12-1,2,3,4,7,8-HxCDF | 100 | 43 | 27-152 | 19-202 | 76-131 |
| 13C12-1,2,3,6,7,8-HxCDF | 100 | 35 | 30-122 | 21-159 | 70-143 |
| 13C12-1,2,3,7,8,9-HxCDF | 100 | 40 | 24-157 | 17-205 | 74-135 |
| 13C12-2,3,4,6,7,8,-HxCDF | 100 | 37 | 29-136 | 22-176 | 73-137 |
| 13C12-1,2,3,4,6,7,8-HpCDD | 100 | 35 | 34-129 | 26-166 | 72-138 |
| 13C12-1,2,3,4,6,7,8-HpCDF | 100 | 41 | 32-110 | 21-158 | 78-129 |
| 13C12-1,2,3,4,7,8,9-HpCDF | 100 | 40 | 28-141 | 20-186 | 77-129 |
| 13C12-OCDD | 200 | 95 | 41-276 | 26-397 | 96-415 |
| 37Cl4-2,3,7,8-TCDD | 10 | 3.6 | 3.9-15.4 | 3.1-19.1 | 7.9-12.7 |
1 All specifications are given as concentration in the final extract, assuming a 20 µL volume.
2 s = standard deviation of the concentration.
3 X = average concentration.
Table 6a - Acceptance Criteria for Performance Tests When Only Tetra Compounds are Tested 1
| CDD/CDF | Test Conc. (ng/mL) | IPR 2 3 | OPR (ng/mL) |
VER (ng/mL) |
|
|---|---|---|---|---|---|
| s (ng/mL) | X (ng/mL) | ||||
| 2,3,7,8-TCDD | 10 | 2.7 | 8.7-12.4 | 7.314.6 | 8.2-12.3 |
| 2,3,7,8-TCDF | 10 | 2.0 | 9.1-13.1 | 8.0-14.7 | 8.6-11.6 |
| 13C12-2,3,7,8-TCDD | 100 | 35 | 32-115 | 25-141 | 85-117 |
| 13C12-2,3,7,8-TCDF | 100 | 34 | 35-99 | 26-126 | 76-131 |
| 37Cl4-2,3,7,8-TCDD | 10 | 3.4 | 4.5-13.4 | 3.7-15.8 | 8.3-12.1 |
1 All specifications are given as concentration in the final extract, assuming a 20 µL volume.
2 s = standard deviation of the concentration.
3 X = average concentration.
Table 7 - Labeled Compounds Recovery in Samples When all CDDS/CDFS are Tested
| Compound | Test conc. (ng/mL) | Labeled
compound recovery |
|
|---|---|---|---|
| (ng/mL) 1 | (%) | ||
| 13C12-2,3,7,8-TCDD | 100 | 25-164 | 25-164 |
| 13C12-2,3,7,8-TCDF | 100 | 24-169 | 24-169 |
| 13C12-1,2,3,7,8-PeCDD | 100 | 25-181 | 25-181 |
| 13C12-1,2,3,7,8-PeCDF | 100 | 24-185 | 24-185 |
| 13C12-2,3,4,7,8-PeCDF | 100 | 21-178 | 21-178 |
| 13C12-1,2,3,4,7,8-HxCDD | 100 | 32-141 | 32-141 |
| 13C12-1,2,3,6,7,8-HxCDD | 100 | 28-130 | 28-130 |
| 13C12-1,2,3,4,7,8-HxCDF | 100 | 26-152 | 26-152 |
| 13C12-1,2,3,6,7,8-HxCDF | 100 | 26-123 | 26-123 |
| 13C12-1,2,3,7,8,9-HxCDF | 100 | 29-147 | 29-147 |
| 13C12-2,3,4,6,7,8-HxCDF | 100 | 28-136 | 28-136 |
| 13C12-1,2,3,4,6,7,8-HpCDD | 100 | 23-140 | 23-140 |
| 13C12-1,2,3,4,6,7,8-HpCDF | 100 | 28-143 | 28-143 |
| 13C12-1,2,3,4,7,8,9-HpCDF | 100 | 26-138 | 26-138 |
| 13C12-OCDD | 200 | 34-313 | 17-157 |
| 37Cl4-2,3,7,8-TCDD | 10 | 3.5-19.7 | 35-197 |
1 Specification given as concentration in the final extract, assuming a 20-µL volume.
Table 7a - Labeled Compound Recovery in Samples When Only Tetra Compounds are Tested
| Compound | Test conc. (ng/mL) | Labeled
compound recovery |
|
|---|---|---|---|
| (ng/mL) 1 | (%) | ||
| 13C12-2,3,7,8-TCDD | 100 | 31-137 | 31-137 |
| 13C12-2,3,7,8-TCDF | 100 | 29-140 | 29-140 |
| 37Cl4-2,3,7,8-TCDD | 10 | 4.2-16.4 | 42-164 |
1 Specification given as concentration in the final extract, assuming a 20 µL volume.
Table 8 - Descriptors, Exact M/Z's, M/Z Types, and Elemental Compositions of the CDDs and CDFs
| Descriptor | Exact M/Z 1 | M/Z type | Elemental composition | Substance 2 |
|---|---|---|---|---|
| 1 | 292.9825 | Lock | C7F11 | PFK |
| 303.9016 | M | C12H4 35Cl4O | TCDF | |
| 305.8987 | M = 2 | C12H4 35Cl3 37ClO | TCDF | |
| 315.9419 | M | 13C12H4 35Cl4O | TCDF 3 | |
| 317.9389 | M = 2 | 13C12H4 35Cl3 37ClO | TCDF 3 | |
| 319.8965 | M | C12H4 35Cl4O2 | TCDD | |
| 321.8936 | M = 2 | C12H4 35Cl3 37ClO2 | TCDD | |
| 327.8847 | M | C12H4 37Cl4O2 | TCDD 4 | |
| 330.9792 | QC | C7F13 | PFK | |
| 331.9368 | M | 13C12H4 35Cl4O2 | TCDD 3 | |
| 333.9339 | M = 2 | 13C12H4 35Cl3 37ClO2 | TCDD 3 | |
| 375.8364 | M = 2 | C12H4 35Cl5 37ClO | HxCDPE | |
| 2 | 339.8597 | M = 2 | C12H3 35Cl4 37ClO | PeCDF |
| 341.8567 | M = 4 | C12H3 35Cl3 37Cl2O | PeCDF | |
| 351.9000 | M = 2 | 13C12H3 35Cl4 37ClO | PeCDF | |
| 353.8970 | M = 4 | 13C12H3 35Cl3 37Cl2O | PeCDF 3 | |
| 354.9792 | Lock | C9F13 | PFK | |
| 355.8546 | M = 2 | C12H3 35Cl4 37ClO2 | PeCDD | |
| 357.8516 | M = 4 | C12H3 35Cl3 37Cl2O2 | PeCDD | |
| 367.8949 | M = 2 | 13C12H3 35Cl4 37ClO2 | PeCDD 3 | |
| 369.8919 | M = 4 | 13C12H3 35Cl3 37Cl2O2 | PeCDD 3 | |
| 409.7974 | M = 2 | C12H3 35Cl6 37ClO | HpCDPE | |
| 3 | 373.8208 | M = 2 | C12H2 35Cl5 37ClO | HxCDF |
| 375.8178 | M = 4 | C12H2 35Cl4 37Cl2O | HxCDF | |
| 383.8639 | M | 13C12H2 35Cl6O | HxCDF 3 | |
| 385.8610 | M = 2 | 13C12H2 35Cl5 37ClO | HxCDF 3 | |
| 389.8157 | M = 2 | C12H2 35Cl5 37ClO2 | HxCDD | |
| 391.8127 | M = 4 | C12H2 35Cl4 37Cl2O2 | HxCDD | |
| 392.9760 | Lock | C9F15 | PFK | |
| 401.8559 | M = 2 | 13C12H2 35Cl5 37ClO2 | HxCDD 3 | |
| 403.8529 | M = 4 | 13C12H2 35Cl4 37Cl2O2 | HxCDD 3 | |
| 430.9729 | QC | C9F17 | PFK | |
| 445.7555 | M = 4 | C12H2 35Cl6 37Cl2O | OCDPE | |
| 4 | 407.7818 | M = 2 | C12H 35Cl6 37ClO | HpCDF |
| 409.7789 | M = 4 | C12H 35Cl5 37Cl2O | HpCDF | |
| 417.8253 | M | 13C12H 35Cl7O | HpCDF 3 | |
| 419.8220 | M = 2 | 13C12H 35Cl6 37ClO | HpCDF 3 | |
| 423.7766 | M = 2 | C12H 35Cl6 37ClO2 | HpCDD | |
| 425.7737 | M = 4 | C12H 35Cl5 37Cl2O2 | HpCDD | |
| 430.9729 | Lock | C9F17 | PFK | |
| 435.8169 | M = 2 | 13C12H 35Cl6 37ClO2 | HpCDD 3 | |
| 437.8140 | M = 4 | 13C12H 35Cl5 37Cl2O2 | HpCDD 3 | |
| 479.7165 | M = 4 | C12H 35Cl7 37Cl2O | NCDPE | |
| 5 | 441.7428 | M = 2 | C12 35Cl7 37ClO | OCDF |
| 442.9728 | Lock | C10F17 | PFK | |
| 443.7399 | M = 4 | C12 35Cl6 37Cl2O | OCDF | |
| 457.7377 | M = 2 | C12 35Cl7 37ClO2 | OCDD | |
| 459.7348 | M = 4 | C12 35Cl6 37Cl2O2 | OCDD | |
| 469.7779 | M = 2 | 13C12 35Cl7 37ClO2 | OCDD 3 | |
| 471.7750 | M = 4 | 13C12 35Cl6 37Cl2O2 | OCDD 3 | |
| 513.6775 | M = 4 | C12 35Cl8 37Cl2O | DCDPE |
1 Nuclidic masses used:
H = 1.007825.
O = 15.994915.
C = 12.00000.
35Cl = 34.968853.
13C = 13.003355.
37Cl = 36.965903.
F = 18.9984.
2 TCDD = Tetrachlorodibenzo-p-dioxin.
PeCDD = Pentachlorodibenzo-p-dioxin.
HxCDD = Hexachlorodibenzo-p-dioxin.
HpCDD = Heptachlorodibenzo-p-dioxin.
OCDD = Octachlorodibenzo-p-dioxin.
HxCDPE = Hexachlorodiphenyl ether.
OCDPE = Octachlorodiphenyl ether.
DCDPE = Decachlorodiphenyl ether.
TCDF = Tetrachlorodibenzofuran.
PeCDF = Pentachlorodibenzofuran.
HxCDF = Hexachlorodibenzofuran.
HpCDF = Heptachlorodibenzofuran.
OCDF = Octachlorodibenzofuran.
HpCDPE = Heptachlorodiphenyl ether.
NCDPE = Nonachlorodiphenyl ether.
PFK = Perfluorokerosene.
3 Labeled compound.
4 There is only one m/z for 37Cl4-2,3,7,8,-TCDD (cleanup standard).
Table 9 - Theoretical Ion Abundance Ratios and QC Limits
| Number of chlorine atoms | M/Z's forming ratio | Theoretical ratio | QC limit 1 | |
|---|---|---|---|---|
| Lower | Upper | |||
| 4 2 | M/(M = 2) | 0.77 | 0.65 | 0.89 |
| 5 | (M = 2)/(M = 4) | 1.55 | 1.32 | 1.78 |
| 6 | (M = 2)/(M = 4) | 1.24 | 1.05 | 1.43 |
| 6 3 | M/(M = 2) | 0.51 | 0.43 | 0.59 |
| 7 | (M = 2)/(M = 4) | 1.05 | 0.88 | 1.20 |
| 7 4 | M/(M = 2) | 0.44 | 0.37 | 0.51 |
| 8 | (M = 2)/(M = 4) | 0.89 | 0.76 | 1.02 |
1 QC limits represent ±15% windows around the theoretical ion abundance ratios.
2 Does not apply to 37Cl4-2,3,7,8-TCDD (cleanup standard).
3 Used for 13C12-HxCDF only.
4 Used for 13C12-HpCDF only.
Table 10 - Suggested Sample Quantities To Be Extracted for Various Matrices 1
| Sample Matrix 2 | Example | Percent solids | Phase | Quantity extracted |
|---|---|---|---|---|
| Single-phase: | ||||
| Aqueous | Drinking water | <1 | ( 3) | 1000 mL. |
| Groundwater | ||||
| Treated wastewater | ||||
| Solid | Dry soil | >20 | Solid | 10 g. |
| Compost | ||||
| Ash | ||||
| Organic | Waste solvent | <1 | Organic | 10 g. |
| Waste oil | ||||
| Organic polymer | ||||
| Tissue | Fish | Organic | 10 g. | |
| Human adipose | ||||
| Multi-phase: | ||||
| Liquid/Solid: | ||||
| Aqueous/Solid | Wet soil | 1-30 | Solid | 10 g. |
| Untreated effluent | ||||
| Digested municipal sludge | ||||
| Filter cake | ||||
| Paper pulp | ||||
| Organic/solid | Industrial sludge | 1-100 | Both | 10 g. |
| Oily waste | ||||
| Liquid/Liquid: | ||||
| Aqueous/organic | In-process effluent | <1 | Organic | 10 g. |
| Untreated effluent | ||||
| Drum waste | ||||
| Aqueous/organic/solid | Untreated effluent | >1 | Organic and solid | 10 g. |
| Drum waste |
1 The quantity of sample to be extracted is adjusted to provide 10 g of solids (dry weight). One liter of aqueous samples containing 1% solids will contain 10 g of solids. For aqueous samples containing greater than 1% solids, a lesser volume is used so that 10 g of solids (dry weight) will be extracted.
2 The sample matrix may be amorphous for some samples. In general, when the CDDs/CDFs are in contact with a multiphase system in which one of the phases is water, they will be preferentially dispersed in or adsorbed on the alternate phase because of their low solubility in water.
3 Aqueous samples are filtered after spiking with the labeled compounds. The filtrate and the materials trapped on the filter are extracted separately, and the extracts are combined for cleanup and analysis.
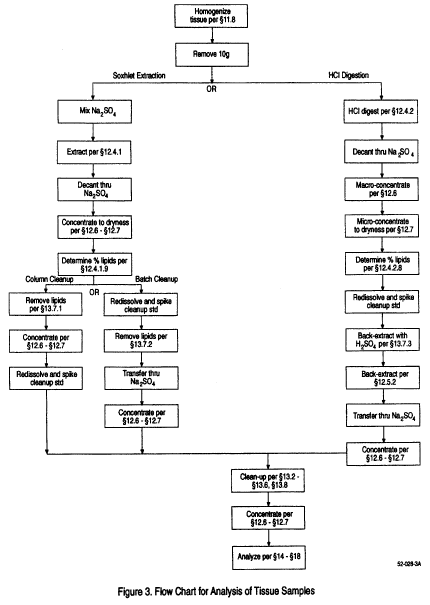

 24.0 Glossary of
Definitions and Purposes
24.0 Glossary of
Definitions and Purposes
These definitions and purposes are specific to this method but have been conformed to common usage as much as possible.
24.1 Units of weight and Measure and Their Abbreviations.
24.1.1 Symbols:
°C - degrees Celsius µL - microliter µm - micrometer < - less than > - greater than % - percent 24.1.2 Alphabetical abbreviations: amp - ampere cm - centimeter g - gram h - hour D - inside diameter in. - inch L - liter M - Molecular ion m - meter mg - milligram min - minute mL - milliliter mm - millimeter m/z - mass-to-charge ratio N - normal; gram molecular weight of solute divided by hydrogen equivalent of solute, per liter of solution OD - outside diameter pg - picogram ppb - part-per-billion ppm - part-per-million ppq - part-per-quadrillion ppt - part-per-trillion psig - pounds-per-square inch gauge v/v - volume per unit volume w/v - weight per unit volume24.2 Definitions and Acronyms (in Alphabetical Order).
Analyte - A CDD or CDF tested for by this method. The analytes are listed in Table 1.
Calibration Standard (CAL) - A solution prepared from a secondary standard and/or stock solutions and used to calibrate the response of the instrument with respect to analyte concentration.
Calibration Verification Standard (VER) - The mid-point calibration standard (CS3) that is used in to verify calibration. See Table 4.
CDD - Chlorinated Dibenzo-p-ioxin - The isomers and congeners of tetra-through octa-chlorodibenzo-p-dioxin.
CDF - Chlorinated Dibenzofuran - The isomers and congeners of tetra-through octa-chlorodibenzofuran.
CS1, CS2, CS3, CS4, CS5 - See Calibration standards and Table 4.
Field Blank - An aliquot of reagent water or other reference matrix that is placed in a sample container in the laboratory or the field, and treated as a sample in all respects, including exposure to sampling site conditions, storage, preservation, and all analytical procedures. The purpose of the field blank is to determine if the field or sample transporting procedures and environments have contaminated the sample.
GC - Gas chromatograph or gas chromatography.
GPC - Gel permeation chromatograph or gel permeation chromatography.
HPLC - High performance liquid chromatograph or high performance liquid chromatography.
HRGC - High resolution GC.
HRMS - High resolution MS.
IPR - Initial precision and recovery; four aliquots of the diluted PAR standard analyzed to establish the ability to generate acceptable precision and accuracy. An IPR is performed prior to the first time this method is used and any time the method or instrumentation is modified.
K-D - Kuderna-Danish concentrator; a device used to concentrate the analytes in a solvent.
Laboratory Blank - See method blank.
Laboratory Control sample (LCS) - See ongoing precision and recovery standard (OPR).
Laboratory Reagent Blank - See method blank.
May - This action, activity, or procedural step is neither required nor prohibited.
May Not - This action, activity, or procedural step is prohibited.
Method Blank - An aliquot of reagent water that is treated exactly as a sample including exposure to all glassware, equipment, solvents, reagents, internal standards, and surrogates that are used with samples. The method blank is used to determine if analytes or interferences are present in the laboratory environment, the reagents, or the apparatus.
Minimum Level (ML) - The level at which the entire analytical system must give a recognizable signal and acceptable calibration point for the analyte. It is equivalent to the concentration of the lowest calibration standard, assuming that all method-specified sample weights, volumes, and cleanup procedures have been employed.
MS - Mass spectrometer or mass spectrometry.
Must - This action, activity, or procedural step is required.
OPR - Ongoing precision and recovery standard (OPR); a laboratory blank spiked with known quantities of analytes. The OPR is analyzed exactly like a sample. Its purpose is to assure that the results produced by the laboratory remain within the limits specified in this method for precision and recovery.
PAR - Precision and recovery standard; secondary standard that is diluted and spiked to form the IPR and OPR.
PFK - Perfluorokerosene; the mixture of compounds used to calibrate the exact m/z scale in the HRMS.
Preparation Blank - See method blank.
Primary Dilution Standard - A solution containing the specified analytes that is purchased or prepared from stock solutions and diluted as needed to prepare calibration solutions and other solutions.
Quality Control Check Sample (QCS) - A sample containing all or a subset of the analytes at known concentrations. The QCS is obtained from a source external to the laboratory or is prepared from a source of standards different from the source of calibration standards. It is used to check laboratory performance with test materials prepared external to the normal preparation process.
Reagent Water - Water demonstrated to be free from the analytes of interest and potentially interfering substances at the method detection limit for the analyte.
Relative Standard Deviation (RSD) - The standard deviation times 100 divided by the mean. Also termed “coefficient of variation.”
RF - Response factor. See Section 10.6.1.
RR - Relative response. See Section 10.5.2.
RSD - See relative standard deviation.
SDS - Soxhlet/Dean-Stark extractor; an extraction device applied to the extraction of solid and semi-solid materials (Reference 7).
Should - This action, activity, or procedural step is suggested but not required.
SICP - Selected ion current profile; the line described by the signal at an exact m/z.
SPE - Solid-phase extraction; an extraction technique in which an analyte is extracted from an aqueous sample by passage over or through a material capable of reversibly adsorbing the analyte. Also termed liquid-solid extraction.
Stock Solution - A solution containing an analyte that is prepared using a reference material traceable to EPA, the National Institute of Science and Technology (NIST), or a source that will attest to the purity and authenticity of the reference material.
TCDD - Tetrachlorodibenzo-p-dioxin.
TCDF - Tetrachlorodibenzofuran.
VER - See calibration verification standard.
Method 1624 Revision B - Volatile Organic Compounds by Isotope Dilution GC/MS 1. Scope and Application1.1 This method is designed to determine the volatile toxic organic pollutants associated with the 1976 Consent Decree and additional compounds amenable to purge and trap gas chromatography-mass spectrometry (GC/MS).
1.2 The chemical compounds listed in table 1 may be determined in municipal and industrial discharges by this method. The methmd is designed to meet the survey requirements of Effluent Guidelines Division (EGD) and the National Pollutants Discharge Elimination System (NPDES) under 40 CFR 136.1 and 136.5. Any modifications of this method, beyond those expressly permitted, shall be considered as major modifications subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.3 The detection limit of this method is usually dependent on the level of interferences rather than instrumental limitations. The limits in table 2 represent the minimum quantity that can be detected with no interferences present.
1.4 The GC/MS portions of this method are for use only by analysts experienced with GC/MS or under the close supervision of such qualified persons. Laboratories unfamiliar with the analyses of environmental samples by GC/MS should run the performance tests in reference 1 before beginning.
2. Summary of Method2.1 Stable isotopically labeled analogs of the compounds of interest are added to a 5 mL water sample. The sample is purged at 20-25 °C with an inert gas in a specially designed chamber. The volatile organic compounds are transferred from the aqueous phase into the gaseous phase where they are passed into a sorbent column and trapped. After purging is completed, the trap is backflushed and heated rapidly to desorb the compounds into a gas chromatograph (GC). The compounds are separated by the GC and detected by a mass spectrometer (MS) (references 2 and 3). The labeled compounds serve to correct the variability of the analytical technique.
2.2 Identification of a compound (qualitative analysis) is performed by comparing the GC retention time and the background corrected characteristic spectral masses with those of authentic standards.
2.3 Quantitative analysis is performed by GC/MS using extracted ion current profile (EICP) areas. Isotope dilution is used when labeled compounds are available; otherwise, an internal standard method is used.
2.4 Quality is assured through reproducible calibration and testing of the purge and trap and GC/MS systems.
3. Contamination and Interferences3.1 Impurities in the purge gas, organic compounds out-gassing from the plumbing upstream of the trap, and solvent vapors in the laboratory account for the majority of contamination problems. The analytical system is demonstrated to be free from interferences under conditions of the analysis by analyzing blanks initially and with each sample lot (samples analyzed on the same 8 hr shift), as described in Section 8.5.
3.2 Samples can be contaminated by diffusion of volatile organic compounds (particularly methylene chloride) through the bottle seal during shipment and storage. A field blank prepared from reagent water and carried through the sampling and handling protocol serves as a check on such contamination.
3.3 Contamination by carry-over can occur when high level and low level samples are analyzed sequentially. To reduce carry-over, the purging device and sample syringe are rinsed between samples with reagent water. When an unusually concentrated sample is encountered, it is followed by analysis of a reagent water blank to check for carry-over. For samples containing large amounts of water soluble materials, suspended solids, high boiling compounds, or high levels or purgeable compounds, the purge device is washed with soap solution, rinsed with tap and distilled water, and dried in an oven at 100-125 °C. The trap and other parts of the system are also subject to contamination; therefore, frequent bakeout and purging of the entire system may be required.
3.4 Interferences resulting from samples will vary considerably from source to source, depending on the diversity of the industrial complex or municipality being sampled.
4. Safety4.1 The toxicity or carcinogenicity of each compound or reagent used in this method has not been precisely determined; however, each chemical compound should be treated as a potential health hazard. Exposure to these compounds should be reduced to the lowest possible level. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of data handling sheets should also be made available to all personnel involved in these analyses. Additional information on laboratory safety can be found in references 4-6.
4.2 The following compounds covered by this method have been tentatively classified as known or suspected human or mammalian carcinogens: benzene, carbon tetrachloride, chloroform, and vinyl chloride. Primary standards of these toxic compounds should be prepared in a hood, and a NIOSH/MESA approved toxic gas respirator should be worn when high concentrations are handled.
5. Apparatus and Materials5.1 Sample bottles for discrete sampling.
5.1.1 Bottle - 25 to 40 mL with screw cap (Pierce 13075, or equivalent). Detergent wash, rinse with tap and distilled water, and dry at >105 °C for one hr minimum before use.
5.1.2 Septum - Teflon-faced silicone (Pierce 12722, or equivalent), cleaned as above and baked at 100-200 °C, for one hour minimum.
5.2 Purge and trap device - consists of purging device, trap, and desorber. Complete devices are commercially available.
5.2.1 Purging device - designed to accept 5 mL samples with water column at least 3 cm deep. The volume of the gaseous head space between the water and trap shall be less than 15 mL. The purge gas shall be introduced less than 5 mm from the base of the water column and shall pass through the water as bubbles with a diameter less than 3 mm. The purging device shown in Figure 1 meets these criteria.
5.2.2 Trap - 25 to 30 cm × 2.5 mm i.d. minimum, containing the following:
5.2.2.1 Methyl silicone packing - one ±0.2 cm, 3 percent OV-1 on 60/80 mesh Chromosorb W, or equivalent.
5.2.2.2 Porous polymer - 15 ±1.0 cm, Tenax GC (2,6-diphenylene oxide polymer), 60/80 mesh, chromatographic grade, or equivalent.
5.2.2.3 Silica gel - 8 ±1.0 cm, Davison Chemical, 35/60 mesh, grade 15, or equivalent. The trap shown in Figure 2 meets these specifications.
5.2.3 Desorber - shall heat the trap to 175 ±5 °C in 45 seconds or less. The polymer section of the trap shall not exceed 180 °C, and the remaining sections shall not exceed 220 °C. The desorber shown in Figure 2 meets these specifications.
5.2.4 The purge and trap device may be a separate unit or coupled to a GC as shown in Figures 3 and 4.
5.3 Gas chromatograph - shall be linearly temperature programmable with initial and final holds, shall contain a glass jet separator as the MS interface, and shall produce results which meet the calibration (Section 7), quality assurance (Section 8), and performance tests (Section 11) of this method.
5.3.1 Column - 2.8 ±0.4 m × 2 ±0.5 mm i. d. glass, packekd with one percent SP-1000 on Carbopak B, 60/80 mesh, or equivalent.
5.4 Mass spectrometer - 70 eV electron impact ionization; shall repetitively scan from 20 to 250 amu every 2-3 seconds, and produce a unit resolution (valleys between m/z 174-176 less than 10 percent of the height of the m/z 175 peak), background corrected mass spectrum from 50 ng 4-bromo-fluorobenzene (BFB) injected into the GC. The BFB spectrum shall meet the mass-intensity criteria in Table 3. All portions of the GC column, transfer lines, and separator which connect the GC column to the ion source shall remain at or above the column temperature during analysis to preclude condensation of less volatile compounds.
5.5 Data system - shall collect and record MS data, store mass intensity data in spectral libraries, process GC/MS data and generate reports, and shall calculate and record response factors.
5.5.1 Data acquisition - mass spectra shall be collected continuously throughout the analysis and stored on a mass storage device.
5.5.2 Mass spectral libraries - user created libraries containing mass spectra obtained from analysis of authentic standards shall be employed to reverse search GC/MS runs for the compounds of interest (Section 7.2).
5.5.3 Data processing - the data system shall be used to search, locate, identify, and quantify the compounds of interest in each GC/MS analysis. Software routines shall be employed to compute retention times and EICP areas. Displays of spectra, mass chromatograms, and library comparisons are required to verify results.
5.5.4 Response factors and multipoint calibrations - the data system shall be used to record and maintain lists of response factors (response ratios for isotope dilution) and generate multi-point calibration curves (Section 7). Computations of relative standard deviation (coefficient of variation) are useful for testing calibration linearity. Statistics on initial and on-going performance shall be maintained (Sections 8 and 11).
5.6 Syringes - 5 mL glass hypodermic, with Luer-lok tips.
5.7 Micro syringes - 10, 25, and 100 uL.
5.8 Syringe valves - 2-way, with Luer ends (Telfon or Kel-F).
5.9 Syringe - 5 mL, gas-tight, with shut-off valve.
5.10 Bottles - 15 mL., screw-cap with Telfon liner.
5.11 Balance - analytical, capable of weighing 0.1 mg.
6. Reagents and Standards6.1 Reagent water - water in which the compounds of interest and interfering compounds are not detected by this method (Section 11.7). It may be generated by any of the following methods:
6.1.1 Activated carbon - pass tap water through a carbon bed (Calgon Filtrasorb-300, or equivalent).
6.1.2 Water purifier - pass tap water through a purifier (Millipore Super Q, or equivalent).
6.1.3 Boil and purge - heat tap water to 90-100 °C and bubble contaminant free inert gas through it for approx one hour. While still hot, transfer the water to screw-cap bottles and seal with a Teflon-lined cap.
6.2 Sodium thiosulfate - ACS granular.
6.3 Methanol - pesticide quality or equivalent.
6.4 Standard solutions - purchased as solution or mixtures with certification to their purity, concentration, and authenticity, or prepared from materials of known purity and composition. If compound purity is 96 percent or greater, the weight may be used without correction to calculate the concentration of the standard.
6.5 Preparation of stock solutions - prepare in methanol using liquid or gaseous standards per the steps below. Observe the safety precautions given in Section 4.
6.5.1 Place approx 9.8 mL of methanol in a 10 mL ground glass stoppered volumetric flask. Allow the flask to stand unstoppered for approximately 10 minutes or until all methanol wetted surfaces have dried. In each case, weigh the flask, immediately add the compound, then immediately reweigh to prevent evaporation losses from affecting the measurement.
6.5.1.1 Liquids - using a 100 µL syringe, permit 2 drops of liquid to fall into the methanol without contacting the leck of the flask. Alternatively, inject a known volume of the compound into the methanol in the flask using a micro-syringe.
6.5.1.2 Gases (chloromethane, bromomethane, chloroethane, vinyl chloride) - fill a valved 5 mL gas-tight syringe with the compound. Lower the needle to approximately 5 mm above the methanol meniscus. Slowly introduce the compound above the surface of the meniscus. The gas will dissolve rapidly in the methanol.
6.5.2 Fill the flask to volume, stopper, then mix by inverting several times. Calculate the concentration in mg/mL (µg/µL) from the weight gain (or density if a known volume was injected).
6.5.3 Transfer the stock solution to a Teflon sealed screw-cap-bottle. Store, with minimal headspace, in the dark at −10 to −20 °C.
6.5.4 Prepare fresh standards weekly for the gases and 2-chloroethylvinyl ether. All other standards are replaced after one month, or sooner if comparison with check standards indicate a change in concentration. Quality control check standards that can be used to determine the accuracy of calibration standards are available from the US Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio.
6.6 Labeled compound spiking solution - from stock standard solutions prepared as above, or from mixtures, prepare the spiking solution to contain a concentration such that a 5-10 µL spike into each 5 mL sample, blank, or aqueous standard analyzed will result in a concentration of 20 µg/L of each labeled compound. For the gases and for the water soluble compounds (acrolein, acrylonitrile, acetone, diethyl ether, and MEK), a concentration of 100 µg/L may be used. Include the internal standards (Section 7.5) in this solution so that a concentration of 20 µg/L in each sample, blank, or aqueous standard will be produced.
6.7 Secondary standards - using stock solutions, prepare a secondary standard in methanol to contain each pollutant at a concentration of 500 µg/mL For the gases and water soluble compounds (Section 6.6), a concentration of 2.5 mg/mL may be used.
6.7.1 Aqueous calibration standards - using a 25 µL syringe, add 20 µL of the secondary standard (Section 6.7) to 50, 100, 200, 500, and 1000 mL of reagent water to produce concentrations of 200, 100, 50, 20, and 10 µg/L, respectively. If the higher concentration standard for the gases and water soluble compounds was chosen (Section 6.6), these compounds will be at concentrations of 1000, 500, 250, 100, and 50 µg/L in the aqueous calibration standards.
6.7.2 Aqueous performance standard - an aqueous standard containing all pollutants, internal standards, labeled compounds, and BFB is prepared daily, and analyzed each shift to demonstrate performance (Section 11). This standard shall contain either 20 or 100 µg/L of the labeled and pollutant gases and water soluble compounds, 10 µg/L BFB, and 20 µg/L of all other pollutants, labeled compounds, and internal standards. It may be the nominal 20 µg/L aqueous calibration standard (Section 6.7.1).
6.7.3 A methanolic standard containing all pollutants and internal standards is prepared to demonstrate recovery of these compounds when syringe injection and purge and trap analyses are compared. This standard shall contain either 100 µg/mL or 500 µg/mL of the gases and water soluble compounds, and 100 µg/mL of the remaining pollutants and internal standards (consistent with the amounts in the aqueous performance standard in 6.7.2).
6.7.4 Other standards which may be needed are those for test of BFB performance (Section 7.1) and for collection of mass spectra for storage in spectral libraries (Section 7.2).
7. Calibration7.1 Assemble the gas chromatographic apparatus and establish operating conditions given in table 2. By injecting standards into the GC, demonstrate that the analytical system meets the detection limits in table 2 and the mass-intensity criteria in table 3 for 50 ng BFB.
7.2 Mass spectral libraries - detection and identification of the compound of interest are dependent upon the spectra stored in user created libraries.
7.2.1 Obtain a mass spectrum of each pollutant and labeled compound and each internal standard by analyzing an authentic standard either singly or as part of a mixture in which there is no interference between closely eluted components. That only a single compound is present is determined by examination of the spectrum. Fragments not attributable to the compound under study indicate the presence of an interfering compound. Adjust the analytical conditions and scan rate (for this test only) to produce an undistorted spectrum at the GC peak maximum. An undistorted spectrum will usually be obtained if five complete spectra are collected across the upper half of the GC peak. Software algorithms designed to “enhance” the spectrum may eliminate distortion, but may also eliminate authentic m/z's or introduce other distortion.
7.2.3 The authentic reference spectrum is obtained under BFB tuning conditions (Section 7.1 and table 3) to normalize it to spectra from other instruments.
7.2.4 The spectrum is edited by saving the 5 most intense mass spectral peaks and all other mass spectral peaks greater than 10 percent of the base peak. This spectrum is stored for reverse search and for compound confirmation.
7.3 Assemble the purge and trap device. Pack the trap as shown in Figure 2 and condition overnight at 170-180 °C by backflushing with an inert gas at a flow rate of 20-30 mL/min. Condition traps daily for a minimum of 10 minutes prior to use.
7.3.1 Analyze the aqueous performance standard (Section 6.7.2) according to the purge and trap procedure in Section 10. Compute the area at the primary m/z (table 4) for each compound. Compare these areas to those obtained by injecting one µL of the methanolic standard (Section 6.7.3) to determine compound recovery. The recovery shall be greater than 20 percent for the water soluble compounds, and 60-110 percent for all other compounds. This recovery is demonstrated initially for each purge and trap GC/MS system. The test is repeated only if the purge and trap or GC/MS systems are modified in any way that might result in a change in recovery.
7.3.2 Demonstrate that 100 ng toluene (or toluene-d8) produces an area at m/z 91 (or 99) approx one-tenth that required to exceed the linear range of the system. The exact value must be determined by experience for each instrument. It is used to match the calibration range of the instrument to the analytical range and detection limits required.
7.4 Calibration by isotope dilution - the isotope dilution approach is used for the purgeable organic compounds when appropriate labeled compounds are available and when interferences do not preclude the analysis. If labeled compounds are not available, or interferences are present, internal standard methods (Section 7.5 or 7.6) are used. A calibration curve encompassing the concentration range of interest is prepared for each compound determined. The relative response (RR) vs concentration (µg/L) is plotted or computed using a linear regression. An example of a calibration curve for toluene using toluene-d8 is given in figure 5. Also shown are the ±10 percent error limits (dotted lines). Relative response is determined according to the procedures described below. A minimum of five data points are required for calibration (Section 7.4.4).
7.4.1 The relative response (RR) of pollutant to labeled compound is determined from isotope ratio values calculated from acquired data. Three isotope ratios are used in this process:
RX = the isotope ratio measured in the pure pollutant (figure 6A). Ry = the isotope ratio of pure labeled compound (figure 6B). Rm = the isotope ratio measured in the analytical mixture of the pollutant and labeled compounds (figure 6C).The correct way to calculate RR is: RR = (Ry−Rm) (RX + 1)/(Rm−RX)(Ry + 1) If Rm is not between 2Ry and 0.5RX, the method does not apply and the sample is analyzed by internal or external standard methods (Section 7.5 or 7.6).
7.4.2 In most cases, the retention times of the pollutant and labeled compound are the same and isotope ratios (R's) can be calculated from the EICP areas, where: R = (area at m1/z)/(area at m2/z) If either of the areas is zero, it is assigned a value of one in the calculations; that is, if: area of m1/z = 50721, and area of m2/z = 0, then R = 50721/1 = 50720. The m/z's are always selected such that RX>Ry. When there is a difference in retention times (RT) between the pollutant and labeled compounds, special precautions are required to determine the isotope ratios.
RX, Ry, and Rm are defined as follows:
RX=[area m1/z (at RT1)]/1 Ry = 1/[area m2/z (at RT2)] Rm=[area m1/z (at RT1)]/[area m2/z (at RT2)]7.4.3 An example of the above calculations can be taken from the data plotted in figure 6 for toluene and toluene-d8. For these data, RX = 168920/1 = 168900, Ry = 1/60960 = 0.00001640, and Rm = 96868/82508 = 1.174. The RR for the above data is then calculated using the equation given in Section 7.4.1. For the example, RR = 1.174.
Note:Not all labeled compounds elute before their pollutant analogs.
7.4.4 To calibrate the analytical system by isotope dilution, analyze a 5 mL aliquot of each of the aqueous calibration standards (Section 6.7.1) spiked with an appropriate constant amount of the labeled compound spiking solution (Section 6.6), using the purge and trap procedure in section 10. Compute the RR at each concentration.
7.4.5 Linearity - if the ratio of relative response to concentration for any compound is constant (less than 20 percent coefficient of variation) over the 5 point calibration range, an averaged relative response/concentration ratio may be used for that compound; otherwise, the complete calibration curve for that compound shall be used over the 5 point calibration range.
7.5 Calibration by internal standard - used when criteria for isotope dilution (Section 7.4) cannot be met. The method is applied to pollutants having no labeled analog and to the labeled compounds. The internal standards used for volatiles analyses are bromochloromethane, 2-bromo-1-chloropropane, and 1,4-dichlorobutane. Concentrations of the labeled compounds and pollutants without labeled analogs are computed relative to the nearest eluted internal standard, as shown in table 2.
7.5.1 Response factors - calibration requires the determination of response factors (RF) which are defined by the following equation:
RF = (AsxCis)/(AisxCs), where As is the EICP area at the characteristic m/z for the compound in the daily standard. Ais is the EICP area at the characteristic m/z for the internal standard.
Cis is the concentration (ug/L) of the internal standard
Cs is the concentration of the pollutant in the daily standard.
7.5.2 The response factor is determined at 10, 20, 50, 100, and 200 ug/L for the pollutants (optionally at five times these concentrations for gases and water soluble pollutants - see Section 6.7), in a way analogous to that for calibration by isotope dilution (Section 7.4.4). The RF is plotted against concentration for each compound in the standard (Cs) to produce a calibration curve.
7.5.3 Linearity - if the response factor (RF) for any compound is constant (less than 35 percent coefficient of variation) over the 5 point calibration range, an averaged response factor may be used for that compound; otherwise, the complete calibration curve for that compound shall be used over the 5 point range.
7.6 Combined calibration - by adding the isotopically labeled compounds and internal standards (Section 6.6) to the aqueous calibration standards (Section 6.7.1), a single set of analyses can be used to produce calibration curves for the isotope dilution and internal standard methods. These curves are verified each shift (Section 11.5) by purging the aqueous performance standard (Section 6.7.2). Recalibration is required only if calibration and on-going performance (Section 11.5) criteria cannot be met.
8. Quality Assurance/Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality assurance program. The minimum requirements of this program consist of an initial demonstration of laboratory capability, analysis of samples spiked with labeled compounds to evaluate and document data quality, and analysis of standards and blanks as tests of continued performance. Laboratory performance is compared to established performance criteria to determine if the results of analyses meet the performance characteristics of the method.
8.1.1 The analyst shall make an initial demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 The analyst is permitted to modify this method to improve separations or lower the costs of measurements, provided all performance specifications are met. Each time a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2 to demonstrate method performance.
8.1.3 Analyses of blanks are required to demonstrate freedom from contamination and that the compounds of interest and interfering compounds have not been carried over from a previous analysis (Section 3). The procedures and criteria for analysis of a blank are described in Sections 8.5 and 11.7.
8.1.4 The laboratory shall spike all samples with labeled compounds to monitor method performance. This test is described in Section 8.3. When results of these spikes indicate atypical method performance for samples, the samples are diluted to bring method performance within acceptable limits (Section 14.2).
8.1.5 The laboratory shall, on an on-going basis, demonstrate through the analysis of the aqueous performance standard (Section 6.7.2) that the analysis system is in control. This procedure is described in Sections 11.1 and 11.5.
8.1.6 The laboratory shall maintain records to define the quality of data that is generated. Development of accuracy statements is described in Sections 8.4 and 11.5.2.
8.2 Initial precision and accuracy - to establish the ability to generate acceptable precision and accuracy, the analyst shall perform the following operations:
8.2.1 Analyze two sets of four 5-mL aliquots (8 aliquots total) of the aqueous performance standard (Section 6.7.2) according to the method beginning in Section 10.
8.2.2 Using results of the first set of four analyses in Section 8.2.1, compute the average recovery (X ) in µg/L and the standard deviation of the recovery (s) in µg/L for each compound, by isotope dilution for polluitants with a labeled analog, and by internal standard for labeled compounds and pollutants with no labeled analog.
8.2.3 For each compound, compare s and X with the corresponding limits for initial precision and accuracy found in table 5. If s and X for all compounds meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may begin. If individual X falls outside the range for accuracy, system performance is unacceptable for that compound.
Note:The large number of compounds in table 5 present a substantial probability that one or more will fail one of the acceptance criteria when all compoulds are analyzed. To determine if the analytical system is out of control, or if the failure can be attributed to probability, proceed as follows:
8.2.4 Using the results of the second set of four analyses, compute s and X for only those compounds which failed the test of the first set of four analyses (Section 8.2.3). If these compounds now pass, system performance is acceptable for all compounds and analysis of blanks and samples may begin. If, however, any of the same compounds fail again, the analysis system is not performing properly for the compound(s) in question. In this event, correct the problem and repeat the entire test (Section 8.2.1).
8.3 The laboratory shall spike all samples with labeled compounds to assess method performance on the sample matrix.
8.3.1 Spike and analyze each sample according to the method beginning in Section 10.
8.3.2 Compute the percent recovery (P) of the labeled compounds using the internal standard method (Section 7.5).
8.3.3 Compare the percent recovery for each compound with the corresponding labeled compound recovery limit in table 5. If the recovery of any compound falls outside its warning limit, method performance is unacceptable for that compound in that sample. Therefore, the sample matrix is complex and the sample is to be diluted and reanalyzed, per Section 14.2.
8.4 As part of the QA program for the laboratory, method accuracy for wastewater samples shall be assessed and records shall be maintained. After the analysis of five wastewater samples for which the labeled compounds pass the tests in Section 8.3.3, compute the average percent recovery (P) and the standard deviation of the percent recovery (sp) for the labeled compounds only. Express the accuracy assessment as a percent recovery interval from P−2sp to P + 2sp. For example, if P = 90% and sp = 10%, the accuracy interval is expressed as 70-110%. Update the accuracy assessment for each compound on a regular basis (e.g. after each 5-10 new accuracy measurements).
8.5 Blanks - reagent water blanks are analyzed to demonstrate freedom from carry-over (Section 3) and contamination.
8.5.1 The level at which the purge and trap system will carry greater than 5 µg/L of a pollutant of interest (table 1) into a succeeding blank shall be determined by analyzing successively larger concentrations of these compounds. When a sample contains this concentration or more, a blank shall be analyzed immediately following this sample to demonstrate no carry-over at the 5 µg/L level.
8.5.2 With each sample lot (samples analyzed on the same 8 hr shift), a blank shall be analyzed immediately after analysis of the aqueous performance standard (Section 11.1) to demonstrate freedom from contamination. If any of the compounds of interest (table 1) or any potentially interfering compound is found in a blank at greater than 10 µg/L (assuming a response factor of 1 relative to the nearest eluted internal standard for compounds not listed in table 1), analysis of samples is halted until the source of contamination is eliminated and a blank shows no evidence of contamination at this level.
8.6 The specifications contained in this method can be met if the apparatus used is calibrated properly, then maintained in a calibrated state.
The standards used for calibration (Section 7), calibration verification (Section 11.5) and for initial (Section 8.2) and on-going (Section 11.5) precision and accuracy should be identical, so that the most precise results will be obtained. The GC/MS instrument in particular will provide the most reproducible results if dedicated to the settings and conditions required for the analyses of volatiles by this method.
8.7 Depending on specific program requirements, field replicates may be collected to determine the precision of the sampling technique, and spiked samples may be required to determine the accuracy of the analysis when internal or external standard methods are used.
9. Sample Collection, Preservation, and Handling9.1 Grab samples are collected in glass containers having a total volume greater than 20 mL. Fill sample bottles so that no air bubbles pass through the sample as the bottle is filled. Seal each bottle so that no air bubbles are entrapped. Maintain the hermetic seal on the sample bottle until time of analysis.
9.2 Samples are maintained at 0-4 °C from the time of collection until analysis. If the sample contains residual chlorine, add sodium thiosulfate preservative (10 mg/40 mL) to the empty sample bottles just prior to shipment to the sample site. EPA Methods 330.4 and 330.5 may be used for measurement of residual chlorine (Reference 8). If preservative has been added, shake bottle vigorously for one minute immediately after filling.
9.3 Experimental evidence indicates that some aromatic compounds, notably benzene, toluene, and ethyl benzene are susceptible to rapid biological degradation under certain environmental conditions. Refrigeration alone may not be adequate to preserve these compounds in wastewaters for more than seven days. For this reason, a separate sample should be collected, acidified, and analyzed when these aromatics are to be determined. Collect about 500 mL of sample in a clean container.
Adjust the pH of the sample to about 2 by adding HCl (1 + 1) while stirring. Check pH with narrow range (1.4 to 2.8) pH paper. Fill a sample container as described in Section 9.1. If residual chlorine is present, add sodium thiosulfate to a separate sample container and fill as in Section 9.1.
9.4 All samples shall be analyzed within 14 days of collection.
10. Purge, Trap, and GC/MS Analysis10.1 Remove standards and samples from cold storage and bring to 20-25 °.
10.2 Adjust the purge gas flow rate to 40 ±4 mL/min. Attach the trap inlet to the purging device and set the valve to the purge mode (figure 3). Open the syringe valve located on the purging device sample introduction needle (figure 1).
10.3 Remove the plunger from a 5-mL syringe and attach a closed syringe valve. Open the sample bottle and carefully pour the sample into the syringe barrel until it overflows. Replace the plunger and compress the sample. Open the syringe valve and vent any residual air while adjusting the sample volume to 5.0 mL. Because this process of taking an aliquot destroys the validity of the sample for future analysis, fill a second syringe at this time to protect against possible loss of data. Add an appropriate amount of the labeled compound spiking solution (Section 6.6) through the valve bore, then close the valve.
10.4 Attach the syringe valve assembly to the syringe valve on the purging device. Open both syringe valves and inject the sample into the purging chamber.
10.5 Close both valves and purge the sample for 11.0 ±0.1 minutes at 20-25 °C.
10.6 After the 11 minute purge time, attach the trap to the chromatograph and set the purge and trap apparatus to the desorb mode (figure 4). Desorb the trapped compounds into the GC column by heating the trap to 170-180 °C while backflushing with carrier gas at 20-60 mL/min for four minutes. Start MS data acquisition upon start of the desorb cycle, and start the GC column temperature program 3 minutes later. Table 1 summarizes the recommended operating conditions for the gas chromatograph. Included in this table are retention times and detection limits that were achieved under these conditions. Other columns may be used provided the requirements in Section 8 can be met. If the priority pollutant gases produce GC peaks so broad that the precision and recovery specifications (Section 8.2) cannot be met, the column may be cooled to ambient or sub-ambient temperatures to sharpen these peaks.
10.7 While analysis of the desorbed compounds proceeds, empty the purging chamber using the sample introduction syringe. Wash the chamber with two 5-mL portions of reagent water. After the purging device has been emptied, allow the purge gas to vent through the chamber until the frit is dry, so that it is ready for the next sample.
10.8 After desorbing the sample for four minutes, recondition the trap by returning to the purge mode. Wait 15 seconds, then close the syringe valve on the purging device to begin gas flow through the trap. Maintain the trap temperature at 170-180 °C. After approximately seven minutes, turn off the trap heater and open the syringe valve to stop the gas flow through the trap. When cool, the trap is ready for the next sample.
11. System Performance11.1 At the beginning of each 8 hr shift during which analyses are performed, system calibration and performance shall be verified for all pollutants and labeled compounds. For these tests, analysis of the aqueous performance standard (Section 6.7.2) shall be used to verify all performance criteria. Adjustment and/or recalibration (per Section 7) shall be performed until all performance criteria are met. Only after all performance criteria are met may blanks and samples be analyzed.
11.2 BFB spectrum validity - the criteria in table 3 shall be met.
11.3 Retention times - the absolute retention times of all compounds shall approximate those given in Table 2.
11.4 GC resolution - the valley height between toluene and toluene-d8 (at m/z 91 and 99 plotted on the same graph) shall be less than 10 percent of the taller of the two peaks.
11.5 Calibration verification and on-going precision and accuracy - compute the concentration of each polutant (Table 1) by isotope dilution (Section 7.4) for those compmunds which have labeled analogs. Compute the concentration of each pollutant (Table 1) which has no labeled analog by the internal standard method (Section 7.5). Compute the concentration of the labeled compounds by the internal standard method. These concentrations are computed based on the calibration data determined in Section 7.
11.5.1 For each pollutant and labeled compound, compare the concentration with the corresponding limit for on-going accuracy in Table 5. If all compmunds meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may continue. If any individual value falls outside the range given, system performance is unacceptable for that compound.
Note:The large number of compounds in Table 5 present a substantial probability that one or more will fail the acceptance criteria when all compounds are analyzed. To determine if the analytical system is out of control, or if the failure may be attributed to probability, proceed as follows:
11.5.1.1 Analyze a second aliquot of the aqueous performance standard (Section 6.7.2).
11.5.1.2 Compute the concentration for only those compounds which failed the first test (Section 11.5.1). If these compounds now pass, system performance is acceptable for all compounds and analyses of blanks and samples may proceed. If, however, any of the compounds fail again, the measurement system is not performing properly for these compounds. In this event, locate and correct the problem or recalibrate the system (Section 7), and repeat the entire test (Section 11.1) for all compounds.
11.5.2 Add results which pass the specification in 11.5.1.2 to initial (Section 8.2) and previous on-going data. Update QC charts to form a graphic representation of laboratory performance (Figure 7). Develop a statement of accuracy for each pollutant and labeled compound by calculating the average percentage recovery (R) and the standard deviation of percent recovery (sr). Express the accuracy as a recovery interval from R−2sr to R + 2sr. For example, if R = 95% and sr = 5%, the accuracy is 85-105 percent.
12. Qualitative Determination - Accomplished by Comparison of Data from Analysis of a Sample or Blank with Data from Analysis of the Shift Standard (Section 11.1). Identification is Confirmed When Spectra and Retention Times Agree Per the Criteria Below12.1 Labeled compounds and pollutants having no labeled analog:
12.1.1 The signals for all characteristic masses stored in the spectral library (Section 7.2.4) shall be present and shall maximize within the same two consecutive scans.
12.1.2 Either (1) the background corrected EICP areas, or (2) the corrected relative intensities of the mass spectral peaks at the GC peak maximum shall agree within a factor of two (0.5 to 2 times) for all masses stored in the library.
12.1.3 The retention time relative to the nearest eluted internal standard shall be within ±7 scans or ±20 seconds, whichever is greater.
12.2 Pollutants having a labeled analog:
12.2.1 The signals for all characteristic masses stored in the spectral library (Section 7.2.4) shall be present and shall maximize within the same two consecutive scans.
12.2.2 Either (1) the background corrected EICP areas, or (2) the corrected relative intensities of the mass spectral peaks at the GC peak maximum shall agree within a factor of two for all masses stored in the spectral library.
12.2.3 The retention time difference between the pollutant and its labeled analog shall agree within ±2 scans or ±6 seconds (whichever is greater) of this difference in the shift standard (Section 11.1).
12.3 Masses present in the experimental mass spectrum that are not present in the reference mass spectrum shall be accounted for by contaminant or background ions. If the experimental mass spectrum is contaminated, an experienced spectrometrist (Section 1.4) is to determine the presence or absence of the compound.
13. Quantitative Determination13.1 Isotope dilution - by adding a known amount of a labeled compound to every sample prior to purging, correction for recovery of the pollutant can be made because the pollutant and its labeled analog exhibit the same effects upon purging, desorption, and gas chromatography. Relative response (RR) values for sample mixtures are used in conjunction with calibration curves described in Section 7.4 to determine concentrations directly, so long as labeled compound spiking levels are constant. For the toluene example given in Figure 6 (Section 7.4.3), RR would be equal to 1.174. For this RR value, the toluene calibration curve given in Figure 5 indicates a concentration of 31.8 µg/L.
13.2 Internal standard - calculate the concentration using the response factor determined from calibration data (Section 7.5) and the following equation:
Concentration = (As × Cis)/(Ais × RF) where the terms are as defined in Section 7.5.1.
13.3 If the EICP area at the quantitation mass for any compound exceeds the calibration range of the system, the sample is diluted by successive factors of 10 and these dilutions are analyzed until the area is within the calibration range.
13.4 Report results for all pollutants and labeled compounds (Table 1) found in all standards, blanks, and samples, in µg/L to three significant figures. Results for samples which have been diluted are reported at the least dilute level at which the area at the quantitation mass is within the calibration range (Section 13.3) and the labeled compound recovery is within the normal range for the Method (Section 14.2).
14. Analysis of Complex Samples14.1 Untreated effluents and other samples frequently contain high levels (>1000 µg/L) of the compounds of interest and of interfering compounds. Some samples will foam excessively when purged; others will overload the trap/or GC column.
14.2 Dilute 0.5 mL of sample with 4.5 mL of reagent water and analyze this diluted sample when labeled compound recovery is outside the range given in Table 5. If the recovery remains outside of the range for this diluted sample, the aqueous performance standard shall be analyzed (Section 11) and calibration verified (Section 11.5). If the recovery for the labeled compmund in the aqueous performance standard is outside the range given in Table 5, the analytical system is out of control. In this case, the instrumelt shall be repaired, the performance specifications in Section 11 shall be met, and the analysis of the undiluted sample shall be repeated. If the recovery for the aqueous performance standard is within the range given in Table 5, the method does not work on the sample being analyzed and the result may not be reported for regulatory compliance purposes.
14.3 Reverse search computer programs can misinterpret the spectrum of chromatographically unresolved pollutant and labeled compound pairs with overlapping spectra when a high level of the pollutant is present. Examine each chromatogram for peaks greater than the height of the internal standard peaks. These peaks can obscure the compounds of interest.
15. Method Performance15.1 The specifications for this method were taken from the inter-laboratory validation of EPA Method 624 (reference 9). Method 1624 has been shown to yield slightly better performance on treated effluents than Method 624. Additional method performance data can be found in Reference 10.
References1. “Performance Tests for the Evaluation of Computerized Gas Chromatography/Mass Spectrometry Equipment and Laboratories,” USEPA, EMSL/Cincinnati, OH 45268, EPA-600/4-80-025 (April 1980).
2. Bellar, T.A. and Lichtenberg, J.J., “Journal American Water Works Association,” 66, 739 (1974).
3. Bellar, T.A. and Lichtenberg, J.J., “Semi-automated Headspace Analysis of Drinking Waters and Industrial Waters for Purgeable Volatile Organic Compounds,” in Measurement of Organic Pollutants Water and Wastewater, C.E. VanHall, ed., American Society for Testing Materials, Philadelphia, PA, Special Technical Publication 686, (1978).
4. “Working with Carcinogens,” DHEW, PHS, NIOSH, Publication 77-206 (1977).
5. “OSHA Safety and Health Standards, General Industry,” 29 CFR part 1910, OSHA 2206, (1976).
6. “Safety in Academic Chemistry Laboratories,” American Chemical Society Publication, Committee on Chemical Safety (1979).
7. “Handbook of Analytical Quality Control in Water and Wastewater Laboratories,” USEPA, EMSL/Cincinnati, OH 45268, EPA-4-79-019 (March 1979).
8. “Methods 330.4 and 330.5 for Total Residual Chlorine,” USEPA, EMSL/Cincinnati, OH 45268, EPA-4-79-020 (March 1979).
9. “EPA Method Study 29 EPA Method 624 - Purgeables,” EPA 600/4-84-054, National Technical Information Service, PB84-209915, Springfield, Virginia 22161, June 1984.
10. “Colby, B.N., Beimer, R.G., Rushneck, D.R., and Telliard, W.A., “Isotope Dilution Gas Chromatography-Mass Spectrometry for the Determination of Priority Pollutants in Industrial Effluents,” USEPA, Effluent Guidelines Division, Washington, DC 20460 (1980).
Table 1 - Volatile Organic Compounds Analyzed by Isotope Dilution Gc/MS
| Compound | Storet | CAS registry | EPA-EGD | NPDES |
|---|---|---|---|---|
| Acetone | 81552 | 67-64-1 | 516 V | |
| Acrolein | 34210 | 107-02-8 | 002 V | 001 V |
| Acrylonitrile | 34215 | 107-13-1 | 003 V | 002 V |
| Benzene | 34030 | 71-43-2 | 004 V | 003 V |
| Bromodichloromethane | 32101 | 75-27-4 | 048 V | 012 V |
| Bromoform | 32104 | 75-25-2 | 047 V | 005 V |
| Bromomethane | 34413 | 74-83-9 | 046 V | 020 V |
| Carbon tetrachloride | 32102 | 56-23-5 | 006 V | 006 V |
| Chlorobenzene | 34301 | 108-90-7 | 007 V | 007 V |
| Chloroethane | 34311 | 75-00-3 | 016 V | 009 V |
| 2-chloroethylvinyl ether | 34576 | 110-75-8 | 019 V | 010 V |
| Chloroform | 32106 | 67-66-1 | 023 V | 011 V |
| Chloromethane | 34418 | 74-87-3 | 045 V | 021 V |
| Dibromochloromethane | 32105 | 124-48-1 | 051 V | 008 V |
| 1,1-dichloroethane | 34496 | 75-34-3 | 013 V | 014 V |
| 1,2-dichloroethane | 34536 | 107-06-2 | 010 V | 015 V |
| 1,1-dichloroethene | 34501 | 75-35-4 | 029 V | 016 V |
| Trans-1,2-dichloroethane | 34546 | 156-60-5 | 030 V | 026 V |
| 1,2-dichloropropane | 34541 | 78-87-5 | 032 V | 017 V |
| Cis-1,3-dichloropropene | 34704 | 10061-01-5 | ||
| Trans-1,3-dichloropropene | 34699 | 10061-02-6 | 033 V | |
| Diethyl ether | 81576 | 60-29-7 | 515 V | |
| P-dioxane | 81582 | 123-91-1 | 527 V | |
| Ethylbenzene | 34371 | 100-41-4 | 038 V | 019 V |
| Methylene chloride | 34423 | 75-09-2 | 044 V | 022 V |
| Methyl ethyl ketone | 81595 | 78-93-3 | 514 V | |
| 1,1,2,2-tetrachloroethane | 34516 | 79-34-5 | 015 V | 023 V |
| Tetrachlorethene | 34475 | 127-18-4 | 085 V | 024 V |
| Toluene | 34010 | 108-88-3 | 086 V | 025 V |
| 1,1,1-trichloroethane | 34506 | 71-55-6 | 011 V | 027 V |
| 1,1,2-trichloroethane | 34511 | 79-00-5 | 014 V | 028 V |
| Trichloroethene | 39180 | 79-01-6 | 087 V | 029 V |
| Vinyl chloride | 39175 | 75-01-4 | 088 V | 031 V |
Table 2 - Gas Chromatography of Purgeable Organic Compounds by Isotope Dilution GC/MS
| EGD No. (1) | Compound | Ref EGD No. | Mean retention time (sec) | Minimum level (2) (µg/L) |
|---|---|---|---|---|
| 181 | Bromochloromethane (I.S.) | 181 | 730 | 10 |
| 245 | Chloromethane-d3 | 181 | 147 | 50 |
| 345 | Chloromethane | 245 | 148 | 50 |
| 246 | Bromomethane-d3 | 181 | 243 | 50 |
| 346 | Bromomethane | 246 | 246 | 50 |
| 288 | Vinyl chloride-d3 | 181 | 301 | 50 |
| 388 | Vinyl chloride | 288 | 304 | 10 |
| 216 | Chloroethane-d5 | 181 | 378 | 50 |
| 316 | Chloroethane | 216 | 386 | 50 |
| 244 | Methylene chloride-d2 | 181 | 512 | 10 |
| 344 | Methylene chloride | 244 | 517 | 10 |
| 616 | Acetone-d6 | 181 | 554 | 50 |
| 716 | Acetone | 616 | 565 | 50 |
| 002 | Acrolein | 181 | 566 | 50 |
| 203 | Acrylonitrile-d3 | 181 | 606 | 50 |
| 303 | Acrylonitrile | 203 | 612 | 50 |
| 229 | 1,1-dichloroethene-d2 | 181 | 696 | 10 |
| 329 | 1,1-dichloroethene | 229 | 696 | 10 |
| 213 | 1,1-dichloroethane-d3 | 181 | 778 | 10 |
| 313 | 1,1-dichloroethane | 213 | 786 | 10 |
| 615 | Diethyl ether-d10 | 181 | 804 | 50 |
| 715 | Diethyl ether | 615 | 820 | 50 |
| 230 | Trans-1,2-dichloroethene-d2 | 181 | 821 | 10 |
| 330 | Trans-1,2-dichloroethene | 230 | 821 | 10 |
| 614 | Methyl ethyl ketone-d3 | 181 | 840 | 50 |
| 714 | Methyl ethyl ketone | 614 | 848 | 50 |
| 223 | Chloroform-13C1 | 181 | 861 | 10 |
| 323 | Chloroform | 223 | 861 | 10 |
| 210 | 1,2-dichloroethane-d4 | 181 | 901 | 10 |
| 310 | 1,2-dichloroethane | 210 | 910 | 10 |
| 211 | 1,1,1-trichloroethane-13C2 | 181 | 989 | 10 |
| 311 | 1,1,1-trichloroethane | 211 | 999 | 10 |
| 527 | p-dioxane | 181 | 1001 | 10 |
| 206 | Carbon tetrachloride-13C1 | 182 | 1018 | 10 |
| 306 | Carbon tetrachloride | 206 | 1018 | 10 |
| 248 | Bromodichloromethane-13C1 | 182 | 1045 | 10 |
| 348 | Bromodichloromethane | 248 | 1045 | 10 |
| 232 | 1,2-dichloropropane-d6 | 182 | 1123 | 10 |
| 332 | 1.2-dichloropropane | 232 | 1134 | 10 |
| 233 | Trans-1,3-dichloropropene-d4 | 182 | 1138 | 10 |
| 333 | Trans-1,3-dichloropropene | 233 | 1138 | 10 |
| 287 | Trichloroethene-13C1 | 182 | 1172 | 10 |
| 387 | Trichloroethene | 287 | 1187 | 10 |
| 204 | Benzene-d6 | 182 | 1200 | 10 |
| 304 | Benzene | 204 | 1212 | 10 |
| 251 | Chlorodibromemethane-13C1 | 182 | 1222 | 10 |
| 351 | Chlorodibromomethane | 251 | 1222 | 10 |
| 214 | 1,1,2-trichloroethane-13C2 | 182 | 1224 | 10 |
| 314 | 1,1,2-trichloroethane | 214 | 1224 | 10 |
| 019 | 2-chloroethylvinyl ether | 182 | 1278 | 10 |
| 182 | 2-bromo-1-chloropropane (I.S.) | 182 | 1306 | 10 |
| 247 | Bromoform-13C1 | 182 | 1386 | 10 |
| 347 | Bromoform | 247 | 1386 | 10 |
| 215 | 1,1,2,2-tetrachloroethane-d2 | 183 | 1525 | 10 |
| 315 | 1,1,2,2-tetrachloroethane | 215 | 1525 | 10 |
| 285 | Tetrachloroethene-13C2 | 183 | 1528 | 10 |
| 385 | Tetrachloroethene | 285 | 1528 | 10 |
| 183 | 1,4-dichlorobutale (int std) | 183 | 1555 | 10 |
| 286 | Toluene-d8 | 183 | 1603 | 10 |
| 386 | Toluene | 286 | 1619 | 10 |
| 207 | Chlorobenzene-d5 | 183 | 1679 | 10 |
| 307 | Chlorobenzene | 207 | 1679 | 10 |
| 238 | Ethylbenzene-d10 | 183 | 1802 | 10 |
| 338 | Ethylbenzene | 238 | 1820 | 10 |
| 185 | Bromofluorobenzene | 183 | 1985 | 10 |
(1) Reference numbers beginning with 0, 1 or 5 indicate a pollutant quantified by the internal standard method; reference numbers beginning with 2 or 6 indicate a labeled compound quantified by the internal standard method; reference numbers beginning with 3 or 7 indicate a pollutant quantified by isotope dilution.
(2) This is a minimum level at which the analytical system shall give recognizable mass spectra (background corrected) and acceptable calibration points. Column: 2.4m (8 ft) × 2 mm i.d. glass, packed with one percent SP-1000 coated on 60/80 Carbopak B. Carrier gas: helium at 40 mL/min. Temperature program: 3 min at 45 °C, 8 °C per min to 240 °C, hold at 240 °C for 15 minutes.
Note: The specifications in this table were developed from data collected from three wastewater laboratories.
Table 3 - BFB Mass-Intensity Specifications
| Mass | Intensity required |
|---|---|
| 50 | 15 to 40 percent of mass 95. |
| 75 | 30 to 60 percent of mass 95. |
| 95 | base peak, 100 percent. |
| 96 | 5 to 9 percent of mass 95. |
| 173 | <2 percent of mass 174. |
| 174 | >50 percent of mass 95. |
| 175 | 5 to 9 percent of mass 174 |
| 176 | 95 to 101 percent of mass 174 |
| 177 | 5 to 9 percent of mass 176. |
Table 4 - Volatile Organic Compound Characteristic Masses
| Labeled compound | Analog | Primary m/z's |
|---|---|---|
| Acetone | d6 | 58/64 |
| Acrolein | d2 | 56/58 |
| Acrylonitrile | d3 | 53/56 |
| Benzene | d6 | 78/84 |
| Bromodichloromethane | 13C | 83/86 |
| Bromoform | 13C | 173/176 |
| Bromomethale | d3 | 96/99 |
| Carbon tetrachloride | 13C | 47/48 |
| Chlorobenzene | d5 | 112/117 |
| Chloroethane | d5 | 64/71 |
| 2-chloroethylvinyl ether | d7 | 106/113 |
| Chloroform | 13C | 85/86 |
| Chloromethane | d3 | 50/53 |
| Dibromochloromethane | 13C | 129/130 |
| 1,1-dichloroethane | d3 | 63/66 |
| 1,2-dichloroethane | d4 | 62/67 |
| 1,1-dichloroethene | d2 | 61/65 |
| Trans-1,2-dichloroethene | d2 | 61/65 |
| 1,2-dichloropropane | d6 | 63/67 |
| Cis-1,3-dichloropropene | d4 | 75/79 |
| Trans-1,3-dichloropropene | d4 | 75/79 |
| Diethyl ether | d10 | 74/84 |
| p-dioxane | d8 | 88/96 |
| Ethylbenzene | d10 | 106/116 |
| Methylene chloride | d2 | 84/88 |
| Methyl ethyl ketone | d3 | 72/75 |
| 1,1,2,2-tetrachloroethane | d2 | 83/84 |
| Tetrachloroethene | 13C2 | 166/172 |
| Toluene | d8 | 92/99 |
| 1,1,1-trichloroethane | d3 | 97/102 |
| 1,1,2-trichloroethane | 13C2 | 83/84 |
| Trichloroethene | 13C | 95/133 |
| Vinyl chloride | d3 | 62/65 |
Table 5 - Acceptance Criteria for Performance Tests
| Compound | Acceptance criteria at 20 µg/L | |||
|---|---|---|---|---|
| Initial precision and accuracy section 8.2.3 | Labeled compound recovery sec. 8.3 and 14.2 | On-going accuracy sec. 11.5 | ||
| s (µg/L) | X (µg/L) | P (percent) | R (µg/L) | |
| Acetone | Note 1 | |||
| Acrolein | Note 2 | |||
| Acrylonitrile | Note 2 | |||
| Benzene | 9.0 | 13.0-28.2 | ns-196 | 4-33 |
| Bromodichloromethane | 8.2 | 6.5-31.5 | ns-199 | 4-34 |
| Bromoform | 7.0 | 7.4-35.1 | ns-214 | 6-36 |
| Bromomethane | 25.0 | d-54.3 | ns-414 | d-61 |
| Carbon tetrachloride | 6.9 | 15.9-24.8 | 42-165 | 12-30 |
| Chlorobenzene | 8.2 | 14.2-29.6 | ns-205 | 4-35 |
| Chloroethane | 14.8 | 2.1-46.7 | ns-308 | d-51 |
| 2-chloroethylvinyl ether | 36.0 | d-69.8 | ns-554 | d-79 |
| Chloroform | 7.9 | 11.6-26.3 | 18-172 | 8-30 |
| Chloromethane | 26.0 | d-55.5 | ns-410 | d-64 |
| Dibromochloromethane | 7.9 | 11.2-29.1 | 16-185 | 8-32 |
| 1,1-dichloroethane | 6.7 | 11.4-31.4 | 23-191 | 9-33 |
| 1,2-dichloroethane | 7.7 | 11.6-30.1 | 12-192 | 8-33 |
| 1,1-dichloroethene | 11.7 | d-49.8 | ns-315 | d-52 |
| Trans-1,2-dichloroethene | 7.4 | 10.5-31.5 | 15-195 | 8-34 |
| 1,2-dichloropropane | 19.2 | d-46.8 | ns-343 | d-51 |
| Cis-1,3-dichloropropene | 22.1 | d-51.0 | ns-381 | d-56 |
| Trans-1,3-dichloropropene | 14.5 | d-40.2 | ns-284 | d-44 |
| Diethyl ether | Note 1 | |||
| P-dioxane | Note 1 | |||
| Ethyl benzene | 9.6 | 15.6-28.5 | ns-203 | 5-35 |
| Methylene chloride | 9.7 | d-49.8 | ns-316 | d-50 |
| Methyl ethyl ketone | Note 1 | |||
| 1,1,2,2-tetrachloroethane | 9.6 | 10.7-30.0 | 5-199 | 7-34 |
| Tetrachloroethene | 6.6 | 15.1-28.5 | 31-181 | 11-32 |
| Toluene | 6.3 | 14.5-28.7 | 4-193 | 6-33 |
| 1,1,1-trichloroethane | 5.9 | 10.5-33.4 | 12-200 | 8-35 |
| 1,1,2-trichloroethane | 7.1 | 11.8-29.7 | 21-184 | 9-32 |
| Trichloroethene | 8.9 | 16.6-29.5 | 35-196 | 12-34 |
| Vinyl chloride | 27.9 | d-58.5 | ns-452 | d-65 |
d = detected; result must be greater than zero.
ns = no specification; limit would be below detection limit.
Note 1: Specifications not available for these compounds at time of release of this method.
Note 2: Specifications not developed for these compounds; use method 603.
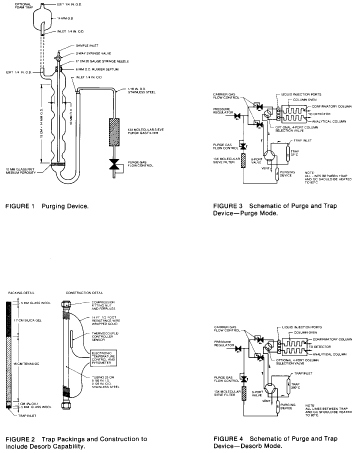
 Method 1625
Revision B - Semivolatile Organic Compounds by Isotope Dilution
GC/MS 1. Scope and Application
Method 1625
Revision B - Semivolatile Organic Compounds by Isotope Dilution
GC/MS 1. Scope and Application
1.1 This method is designed to determine the semivolatile toxic organic pollutants associated with the 1976 Consent Decree and additional compounds amenable to extraction and analysis by capillary column gas chromatography-mass spectrometry (GC/MS).
1.2 The chemical compounds listed in Tables 1 and 2 may be determined in municipal and industrial discharges by this method. The method is designed to meet the survey requirements of Effluent Guidelines Division (EGD) and the National Pollutants Discharge Elimination System (NPDES) under 40 CFR 136.1. Any modifications of this method, beyond those expressly permitted, shall be considered as major modifications subject to application and approval of alternate test procedures under 40 CFR 136.4 and 136.5.
1.3 The detection limit of this method is usually dependent on the level of interferences rather than instrumental limitations. The limits listed in Tables 3 and 4 represent the minimum quantity that can be detected with no interferences present.
1.4 The GC/MS portions of this method are for use only by analysts experienced with GC/MS or under the close supervision of such qualified persons. Laboratories unfamiliar with analyses of environmental samples by GC/MS should run the performance tests in reference 1 before beginning.
2. Summary of Method2.1 Stable isotopically labeled analogs of the compounds of interest are added to a one liter wastewater sample. The sample is extracted at pH 12-13, then at pH <2 with methylene chloride using continuous extraction techniques. The extract is dried over sodium sulfate and concentrated to a volume of one mL. An internal standard is added to the extract, and the extract is injected into the gas chromatograph (GC). The compounds are separated by GC and detected by a mass spectrometer (MS). The labeled compounds serve to correct the variability of the analytical technique.
2.2 Identification of a compound (qualitative analysis) is performed by comparing the GC retention time and background corrected characteristic spectral masses with those of authentic standards.
2.3 Quantitative analysis is performed by GC/MS using extracted ion current profile (EICP) areas. Isotope dilution is used when labeled compounds are available; otherwise, an internal standard method is used.
2.4 Quality is assured through reproducible calibration and testing of the extraction and GC/MS systems.
3. Contamination and Interferences3.1 Solvents, reagents, glassware, and other sample processing hardware may yield artifacts and/or elevated baselines causing misinterpretation of chromatograms and spectra. All materials shall be demonstrated to be free from interferences under the conditions of analysis by running method blanks initially and with each sample lot (samples started through the extraction process on a given 8 hr shift, to a maximum of 20). Specific selection of reagents and purification of solvents by distillation in all-glass systems may be required. Glassware and, where possible, reagents are cleaned by solvent rinse and baking at 450 °C for one hour minimum.
3.2 Interferences coextracted from samples will vary considerably from source to source, depending on the diversity of the industrial complex or municipality being samples.
4. Safety4.1 The toxicity or carcinogenicity of each compound or reagent used in this method has not been precisely determined; however, each chemical compound should be treated as a potential health hazard. Exposure to these compounds should be reduced to the lowest possible level. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of data handling sheets should also be made available to all personnel involved in these analyses. Additional information on laboratory safety can be found in references 2-4.
4.2 The following compounds covered by this method have been tentatively classified as known or suspected human or mammalian carcinogens: benzidine benzo(a)anthracene, 3,3′-dichlorobenzidine, benzo(a)pyrene, dibenzo(a,h)anthracene, N-nitrosodimethylamine, and β-naphtylamine. Primary standards of these compounds shall be prepared in a hood, and a NIOSH/MESA approved toxic gas respirator should be worn when high concentrations are handled.
5. Apparatus and Materials5.1 Sampling equipment for discrete or composite sampling.
5.1.1 Sample bottle, amber glass, 1.1 liters minimum. If amber bottles are not available, samples shall be protected from light. Bottles are detergent water washed, then solvent rinsed or baked at 450 °C for one hour minimum before use.
5.1.2 Bottle caps - threaded to fit sample bottles. Caps are lined with Teflon. Aluminum foil may be substituted if the sample is not corrosive. Liners are detergent water washed, then reagent water (Section 6.5) and solvent rinsed, and baked at approximately 200 °C for one hour minimum before use.
5.1.3 Compositing equipment - automatic or manual compositing system incorporating glass containers for collection of a minimum 1.1 liters. Sample containers are kept at 0 to 4 °C during sampling. Glass or Teflon tubing only shall be used. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used in the pump only. Before use, the tubing is thoroughly rinsed with methanol, followed by repeated rinsings with reagent water (Section 6.5) to minimize sample contamination. An integrating flow meter is used to collect proportional composite samples.
5.2 Continuous liquid-liquid extractor - Teflon or glass conncecting joints and stopcocks without lubrication (Hershberg-Wolf Extractor) one liter capacity, Ace Glass 6841-10, or equivalent.
5.3 Drying column - 15 to 20 mm i.d. Pyrex chromatographic column equipped with coarse glass frit or glass wool plug.
5.4 Kuderna-Danish (K-D) apparatus
5.4.1 Concentrator tube - 10mL, graduated (Kontes K-570050-1025, or equivalent) with calibration verified. Ground glass stopper (size 19/22 joint) is used to prevent evaporation of extracts.
5.4.2 Evaporation flask - 500 mL (Kontes K-570001-0500, or equivalent), attached to concentrator tube with springs (Kontes K-662750-0012).
5.4.3 Snyder column - three ball macro (Kontes K-503000-0232, or equivalent).
5.4.4 Snyder column - two ball micro (Kontes K-469002-0219, or equivalent).
5.4.5 Boiling chips - approx 10/40 mesh, extracted with methylene chloride and baked at 450 °C for one hr minimum.
5.5 Water bath - heated, with concentric ring cover, capable of temperature control ±2 °C, installed in a fume hood.
5.6 Sample vials - amber glass, 2-5 mL with Teflon-lined screw cap.
5.7 Analytical balance - capable of weighing 0.1 mg.
5.8 Gas chromatograph - shall have splitless or on-column injection port for capillary column, temperature program with 30 °C hold, and shall meet all of the performance specifications in Section 12.
5.8.1 Column - 30 ±5 m × 0.25 ±0.02 mm i.d. 5% phenyl, 94% methyl, 1% vinyl silicone bonded phase fused silica capillary column (J & W DB-5, or equivalent).
5.9 Mass spectrometer - 70 eV electron impact ionization, shall repetitively scan from 35 to 450 amu in 0.95 to 1.00 second, and shall produce a unit resolution (valleys between m/z 441-442 less than 10 percent of the height of the 441 peak), backgound corrected mass spectrum from 50 ng decafluorotriphenylphosphine (DFTPP) introduced through the GC inlet. The spectrum shall meet the mass-intensity criteria in Table 5 (reference 5). The mass spectrometer shall be interfaced to the GC such that the end of the capillary column terminates within one centimeter of the ion source but does not intercept the electron or ion beams. All portions of the column which connect the GC to the ion source shall remain at or above the column temperature during analysis to preclude condensation of less volatile compounds.
5.10 Data system - shall collect and record MS data, store mass-intensity data in spectral libraries, process GC/MS data, generate reports, and shall compute and record response factors.
5.10.1 Data acquisition - mass spectra shall be collected continuously throughout the analysis and stored on a mass storage device.
5.10.2 Mass spectral libraries - user created libraries containing mass spectra obtained from analysis of authentic standards shall be employed to reverse search GC/MS runs for the compounds of interest (Section 7.2).
5.10.3 Data processing - the data system shall be used to search, locate, identify, and quantify the compounds of interest in each GC/MS analysis. Software routines shall be employed to compute retention times and peak areas. Displays of spectra, mass chromatograms, and library comparisons are required to verify results.
5.10.4 Response factors and multipoint calibrations - the data system shall be used to record and maintain lists of response factors (response ratios for isotope dilution) and multipoint calibration curves (Section 7). Computations of relative standard deviation (coefficient of variation) are useful for testing calibration linearity. Statistics on initial (Section 8.2) and on-going (Section 12.7) performance shall be computed and maintained.
6. Reagents and Standards6.1 Sodium hydroxide - reagent grade, 6N in reagent water.
6.2 Sulfuric acid - reagent grade, 6N in reagent water.
6.3 Sodium sulfate - reagent grade, granular anhydrous, rinsed with methylene chloride (20 mL/g) and conditioned at 450 °C for one hour minimum.
6.4 Methylene chloride - distilled in glass (Burdick and Jackson, or equivalent).
6.5 Reagent water - water in which the compounds of interest and interfering compounds are not detected by this method.
6.6 Standard solutions - purchased as solutions or mixtures with certification to their purity, concentration, and authenticity, or prepared from materials of known purity and composition. If compound purity is 96 percent or greater, the weight may be used without correction to compute the concentration of the standard. When not being used, standards are stored in the dark at −20 to −10 °C in screw-capped vials with Teflon-lined lids. A mark is placed on the vial at the level of the solution so that solvent evaporation loss can be detected. The vials are brought to room temperature prior to use. Any precipitate is redissolved and solvent is added if solvent loss has occurred.
6.7 Preparation of stock solutions - prepare in methylene chloride, benzene, p-dioxane, or a mixture of these solvents per the steps below. Observe the safety precautions in Section 4. The large number of labeled and unlabeled acid, base/neutral, and Appendix C compounds used for combined calibration (Section 7) and calibration verification (12.5) require high concentratimns (approx 40 mg/mL) when individual stock solutions are prepared, so that dilutions of mixtures will permit calibration with all compounds in a single set of solutions. The working range for most compounds is 10-200 µg/mL. Compounds with a reduced MS response may be prepared at higher concentrations.
6.7.1 Dissolve an appropriate amount of assayed reference material in a suitable solvent. For example, weigh 400 mg naphthalene in a 10 mL ground glass stoppered volumetric flask and fill to the mark with benzene. After the naphthalene is completely dissolved, transfer the solution to a 15 mL vial with Teflon-lined cap.
6.7.2 Stock standard solutions should be checked for signs of degradation prior to the preparation of calibration or performance test standards. Quality control check samples that can be used to determine the accuracy of calibration standards are available from the US Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Cincinnati, Ohio 45268.
6.7.3 Stock standard solutions shall be replaced after six months, or sooner if comparison with quality control check samples indicates a change in concentration.
6.8 Labeled compound spiking solution - from stock standard solutions prepared as above, or from mixtures, prepare the spiking solution at a concentration of 200 µg/mL, or at a concentration appropriate to the MS response of each compound.
6.9 Secondary standard - using stock solutions (Section 6.7), prepare a secondary standard containing all of the compounds in Tables 1 and 2 at a concentration of 400 µg/mL, or higher concentration appropriate to the MS response of the compound.
6.10 Internal standard solution - prepare 2,2′-difluorobiphenyl (DFB) at a concentration of 10 mg/mL in benzene.
6.11 DFTPP solution - prepare at 50 µg/mL in acetone.
6.12 Solutions for obtaining authentic mass spectra (Section 7.2) - prepare mixtures of compounds at concentrations which will assure authentic spectra are obtained for storage in libraries.
6.13 Calibration solutions - combine 0.5 mL of the solution in Section 6.8 with 25, 50, 125, 250, and 500 uL of the solution in section 6.9 and bring to 1.00 mL total volume each. This will produce calibration solutions of nominal 10, 20, 50, 100, and 200 µg/mL of the pollutants and a constant nominal 100 µg/mL of the labeled compounds. Spike each solution with 10 µL of the internal standard solution (Section 6.10). These solutions permit the relative response (labeled to unlabeled) to be measured as a function of concentration (Section 7.4).
6.14 Precision and recovery standard - used for determination of initial (Section 8.2) and on-going (Section 12.7) precision and recovery. This solution shall contain the pollutants and labeled compounds at a nominal concentration of 100 µg/mL.
6.15 Stability of solutions - all standard solutions (Sections 6.8-6.14) shall be analyzed within 48 hours of preparation and on a monthly basis thereafter for signs of degradation. Standards will remain acceptable if the peak area at the quantitation mass relative to the DFB internal standard remains within ±15 percent of the area obtained in the initial analysis of the standard.
7. Calibration7.1 Assemble the GC/MS and establish the operating conditions in Table 3. Analyze standards per the procedure in Section 11 to demonstrate that the analytical system meets the detection limits in Tables 3 and 4, and the mass-intensity criteria in Table 5 for 50 ng DFTPP.
7.2 Mass spectral libraries - detection and identification of compounds of interest are dependent upon spectra stored in user created libraries.
7.2.1 Obtain a mass spectrum of each pollutant, labeled compound, and the internal standard by analyzing an authentic standard either singly or as part of a mixture in which there is no interference between closely eluted components. That only a single compound is present is determined by examination of the spectrum. Fragments not attributable to the compound under study indicate the presence of an interfering compound.
7.2.2 Adjust the analytical conditions and scan rate (for this test only) to produce an undistorted spectrum at the GC peak maximum. An undistorted spectrum will usually be obtained if five complete spectra are collected across the upper half of the GC peak. Software algorithms designed to “enhance” the spectrum may eliminate distortion, but may also eliminate authentic masses or introduce other distortion.
7.2.3 The authentic reference spectrum is obtained under DFTPP tuning conditions (Section 7.1 and Table 5) to normalize it to spectra from other instruments.
7.2.4 The spectrum is edited by saving the 5 most intense mass spectral peaks and all other mass spectral peaks greater than 10 percent of the base peak. This edited spectrum is stored for reverse search and for compound confirmation.
7.3 Analytical range - demonstrate that 20 ng anthracene or phenanthrene produces an area at m/z 178 approx one-tenth that required to exceed the linear range of the system. The exact value must be determined by experience for each instrument. It is used to match the calibration range of the instrument to the analytical range and detection limits required, and to diagnose instrument sensitivity problems (Section 15.4). The 20 ug/mL calibration standard (Section 6.13) can be used to demonstrate this performance.
7.3.1 Polar compound detection - demonstrate that unlabeled pentachlorophenol and benzidine are detectable at the 50 µg/mL level (per all criteria in Section 13). The 50 µg/mL calibration standard (Section 6.13) can be used to demonstrate this performance.
7.4 Calibration with isotope dilution - isotope dilution is used when (1) labeled compounds are available, (2) interferences do not preclude its use, and (3) the quantitation mass extracted ion current profile (EICP) area for the compound is in the calibration range. If any of these conditions preclude isotope dilution, internal standard methods (Section 7.5 or 7.6) are used.
7.4.1 A calibration curve encompassing the concentration range is prepared for each compound to be determined. The relative response (pollutant to labeled) vs concentration in standard solutions is plotted or computed using a linear regression. The example in Figure 1 shows a calibration curve for phenol using phenol-d5 as the isotopic diluent. Also shown are the ±10 percent error limits (dotted lines). Relative Reponse (RR) is determined according to the procedures described below. A minimum of five data points are employed for calibration.
7.4.2 The relative response of a pollutant to its labeled analog is determined from isotope ratio values computed from acquired data. Three isotope ratios are used in this process:
RX = the isotope ratio measured for the pure pollutant.
Ry = the isotope ratio measured for the labeled compound.
Rm = the isotope ratio of an analytical mixture of pollutant and labeled compounds.
The m/z's are selected such that RX>Ry. If Rm is not between 2Ry and 0.5RX, the method does not apply and the sample is analyzed by internal or external standard methods.
7.4.3 Capillary columns usually separate the pollutant-labeled pair, with the labeled compound eluted first (Figure 2). For this case, RX = [area m1/z]/1, at the retention time of the pollutant (RT2). Ry = 1/[area m2/z, at the retention time of the labeled compound RT1). Rm = [area at m1/z (at RT2)]/[area at RT1)], as measured in the mixture of the pollutant and labeled compounds (Figure 2), and RR = Rm.
7.4.4 Special precautions are taken when the pollutant-labeled pair is not separated, or when another labeled compound with interfering spectral masses overlaps the pollutant (a case which can occur with isomeric compounds). In this case, it is necessary to determine the respective contributions of the pollutant and labeled compounds to the respective EICP areas. If the peaks are separated well enough to permit the data system or operator to remove the contributions of the compounds to each other, the equations in Section 7.4.3 apply. This usually occurs when the height of the valley between the two GC peaks at the same m/z is less than 10 percent of the height of the shorter of the two peaks. If significant GC and spectral overlap occur, RR is computed using the following equation:
RR = (Ry − Rm) (RX + 1)/(Rm − RX) (Ry + 1), where RX is measured as shown in Figure 3A, Ry is measured as shown in Figure 3B, and Rm is measured as shown in Figure 3C. For example, RX = 46100/4780 = 9.644, Ry = 2650/43600 = 0.0608, Rm = 49200/48300 = 1.019. amd RR = 1.114.
7.4.5 To calibrate the analytical system by isotope dilution, analyze a 1.0 µL aliquot of each of the calibration standards (Section 6.13) using the procedure in Section 11. Compute the RR at each concentration.
7.4.6 Linearity - if the ratio of relative response to concentration for any compound is constant (less than 20 percent coefficient of variation) over the 5 point calibration range, and averaged relative response/concentration ratio may be used for that compound; otherwise, the complete calibration curve for that compound shall be used over the 5 point calibration range.
7.5 Calibration by internal standard - used when criteria for istope dilution (Section 7.4) cannot be met. The internal standard to be used for both acid and base/neutral analyses is 2,2′-difluorobiphenyl. The internal standard method is also applied to determination of compounds having no labeled analog, and to measurement of labeled compounds for intra-laboratory statistics (Sections 8.4 and 12.7.4).
7.5.1 Response factors - calibration requires the determination of response factors (RF) which are defined by the following equation:
RF = (As × Cis)/(Ais × Cs), where As is the area of the characteristic mass for the compmund in the daily standard Ais is the area of the characteristic mass for the internal standard Cis is the concentration of the internal standard (µg/mL) Cs is the concentration of the compound in the daily standard (µg/mL)7.5.1.1 The response factor is determined for at least five concentrations appropriate to the response of each compound (Section 6.13); nominally, 10, 20, 50, 100, and 200 µg/mL. The amount of internal standard added to each extract is the same (100 µg/mL) so that Cis remains constant. The RF is plotted vs concentration for each compound in the standard (Cs) to produce a calibration curve.
7.5.1.2 Linearity - if the response factor (RF) for any compound is constant (less than 35 percent coefficient of variation) over the 5 point calibration range, an averaged response factor may be used for that compound; otherwise, the complete calibration curve for that compound shall be used over the 5 point range.
7.6 Combined calibration - by using calibration solutions (Section 6.13) containing the pollutants, labeled compounds, and the internal standard, a single set of analyses can be used to produce calibration curves for the isotope dilution and internal standard methods. These curves are verified each shift (Section 12.5) by analyzing the 100 µg/mL calibration standard (Section 6.13). Recalibration is required only if calibration verification (Section 12.5) criteria cannot be met.
8. Quality Assurance/Quality Control8.1 Each laboratory that uses this method is required to operate a formal quality assurance program. The minimum requirements of this program consist of an initial demonstration of laboratory capability, analysis of samples spiked with labeled compounds to evaluate and document data quality, and analysis of standards and blanks as tests of continued performance. Laboratory performance is compared to established performance criteria to determine if the results of analyses meet the performance characteristics of the method.
8.1.1 The analyst shall make an initial demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 8.2.
8.1.2 The analyst is permitted to modify this method to improve separations or lower the costs of measurements, provided all performance specifications are met. Each time a modification is made to the method, the analyst is required to repeat the procedure in Section 8.2 to demonstrate method performance.
8.1.3 Analyses of blanks are required to demonstrate freedom from contamination. The procedures and criteria for analysis of a blank are described in Section 8.5.
8.1.4 The laboratory shall spike all samples with labeled compounds to monitor method performance. This test is described in Section 8.3. When results of these spikes indicate atypical method performance for samples, the samples are diluted to bring method performance within acceptable limits (Section 15).
8.1.5 The laboratory shall, on an on-going basis, demonstrate through calibration verification and the analysis of the precision and recovery standard (Section 6.14) that the analysis system is in control. These procedures are described in Sections 12.1, 12.5, and 12.7.
8.1.6 The laboratory shall maintain records to define the quality of data that is generated. Development of accuracy statements is described in Section 8.4.
8.2 Initial precision and accuracy - to establish the ability to generate acceptable precision and accuracy, the analyst shall perform the following operations:
8.2.1 Extract, concentrate, and analyze two sets of four one-liter aliquots (8 aliquots total) of the precision and recovery standard (Section 6.14) according to the procedure in Section 10.
8.2.2 Using results of the first set of four analyses, compute the average recovery (X ) in µg/mL and the standard deviation of the recovery (s) in θg/µL for each compound, by isotope dilution for pollutants with a labeled analog, and by internal standard for labeled compounds and pollutants with no labeled analog.
8.2.3 For each compound, compare s and X with the corresponding limits for initial precision and accuracy in Table 8. If s and X for all compounds meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may begin. If, however, any individual s exceeds the precision limit or any individual X falls outside the range for accuracy, system performance is unacceptable for that compound.
Note:The large number of compounds in Table 8 present a substantial probability that one or more will fail the acceptance criteria when all compounds are analyzed. To determine if the analytical system is out of control, or if the failure can be attributed to probability, proceed as follows:
8.2.4 Using the results of the second set of four analyses, compute s and X for only those compounds which failed the test of the first set of four analyses (Section 8.2.3). If these compounds now pass, system performance is acceptable for all compounds and analysis of blanks and samples may begin. If, however, any of the same compoulds fail again, the analysis system is not performing properly for these compounds. In this event, correct the problem and repeat the entire test (Section 8.2.1).
8.3 The laboratory shall spike all samples with labeled compounds to assess method performance on the sample matrix.
8.3.1 Analyze each sample according to the method in Section 10.
8.3.2 Compute the percent recovery (P) of the labeled compounds using the internal standard methmd (Section 7.5).
8.3.3 Compare the labeled compound recovery for each compound with the corresponding limits in Table 8. If the recovery of any compounds falls outside its warning limit, method performance is unacceptable for that compound in that sample, Therefore, the sample is complex and is to be diluted and reanalyzed per Section 15.4.
8.4 As part of the QA program for the laboratory, method accuracy for wastewater samples shall be assessed and records shall be maintained. After the analysis of five wastewater samples for which the labeled compounds pass the tests in Section 8.3, compute the average percent recovery (P) and the standard deviation of the percent recovery (sp) for the labeled compounds only. Express the accuracy assessment as a percent recovery interval from P - 2 sp to P + 2sp. For example, if P = 90% and sp = 10%, the accuracy interval is expressed as 70-100%. Update the accuracy assessment for each compound on a regular basis (e.g. after each 5-10 new accuracy measurements).
8.5 Blanks - reagent water blanks are analyzed to demonstrate freedom from contamination.
8.5.1 Extract and concentrate a blank with each sample lot (samples started through the extraction process on the same 8 hr shift, to a maximum of 20 samples). Analyze the blank immediately after analysis of the precision and recovery standard (Section 6.14) to demonstrate freedom from contamination.
8.5.2 If any of the compounds of interest (Tables 1 and 2) or any potentially interfering compound is found in a blank at greater than 10 µg/L (assuming a response factor of 1 relative to the internal standard for compounds not listed in Tables 1 and 2), analysis of samples is halted until the source of contamination is eliminated and a blank shows no evidence of contamination at this level.
8.6 The specifications contained in this method can be met if the apparatus used is calibrated properly, then maintained in a calibrated state. The standards used for calibration (Section 7), calibration verification (Section 12.5), and for initial (Section 8.2) and on-going (Section 12.7) precision and recovery should be identical, so that the most precise results will be obtained. The GC/MS instrument in particular will provide the most reproducible results if dedicated to the settings and conditions required for the analysis of semi-volatiles by this method.
8.7 Depending on specific program requirements, field replicates may be collected to determine the precision of the sampling technique, and spiked samples may be required to determine the accuracy of the analysis when internal or external standard methods are used.
9. Sample Collection, Preservation, and Handling9.1 Collect samples in glass containers following conventional sampling practices (Reference 7). Composite samples are collected in refrigerated glass containers (Section 5.1.3) in accordance with the requirements of the sampling program.
9.2 Maintain samples at 0-4 °C from the time collectimn until extraction. If residual chlorine is present, add 80 mg sodium thiosulfate per liter of water. EPA Methods 330.4 and 330.5 may be used to measure residual chlorine (Reference 8).
9.3 Begin sample extraction within seven days of collection, and analyze all extracts within 40 days of extraction.
10. Sample Extraction and Concentration (See Figure 4)10.1 Labeled compound spiking - measure 1.00 ±0.01 liter of sample into a glass container. For untreated effluents, and samples which are expected to be difficult to extract and/or concentrate, measure an additional 10.0 ±0.1 mL and dilute to a final volume of 1.00 ±0.01 liter with reagent water in a glass container.
10.1.1 For each sample or sample lot (to a maximum of 20) to be extracted at the same time, place three 1.00 ±0.10 liter aliquots of reagent water in glass containers.
10.1.2 Spike 0.5 mL of the labeled compound spiking solution (Section 6.8) into all samples and one reagant water aliquot.
10.1.3 Spike 1.0 mL of the precision and recovery standard (Section 6.14) into the two remaining reagent water aliquots.
10.1.4 Stir and equilibrate all solutions for 1-2 hr.
10.2 Base/neutral extraction - place 100-150 mL methylene chloride in each continuous extractor and 200-300 in each distilling flask.
10.2.1 Pour the sample(s), blank, and standard aliquots into the extractors. Rinse the glass containers with 50-100 mL methylene chloride and add to the respective extractor.
10.2.2 Adjust the pH of the waters in the extractors to 12-13 with 6N NaOH while monitoring with a pH meter. Begin the extraction by heating the flask until the methylene chloride is boiling. When properly adjusted, 1-2 drops of methylene chloride per second will fall from the condensor tip into the water. After 1-2 hours of extraction, test the pH and readjust to 12-13 if required. Extract for 18-24 hours.
10.2.3 Remove the distilling flask, estimate and record the volume of extract (to the nearest 100 mL), and pour the contents through a drying column containing 7 to 10 cm anhydrous sodium sulfate. Rinse the distilling flask with 30-50 mL of methylene chloride and pour through the drying column. Collect the solution in a 500 mL K-D evaporator flask equipped with a 10 mL concentrator tube. Seal, label as the base/neutral fraction, and concentrate per Sections 10.4 to 10.5.
10.3 Acid extraction - adjust the pH of the waters in the extractors to 2 or less using 6N sulfuric acid. Charge clean distilling flasks with 300-400 mL of methylene chloride. Test and adjust the pH of the waters after the first 1-2 hr of extraction. Extract for 18-24 hours.
10.3.1 Repeat Section 10.2.3, except label as the acid fraction.
10.4 Concentration - concentrate the extracts in separate 500 mL K-D flasks equipped with 10 mL concentrator tubes.
10.4.1 Add 1 to 2 clean boiling chips to the flask and attach a three-ball macro Snyder column. Prewet the column by adding approximately one mL of methylene chloride through the top. Place the K-D apparatus in a hot water bath so that the entire lower rounded surface of the flask is bathed with steam. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 minutes. At the proper rate of distillation, the balls of the column will actively chatter but the chambers will not flood. When the liquid has reached an apparent volume of 1 mL, remove the K-D apparatus from the bath and allow the solvent to drain and cool for at least 10 minutes. Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1-2 mL of methylene chloride. A 5-mL syringe is recommended for this operation.
10.4.2 For performance standards (Sections 8.2 and 12.7) and for blanks (Section 8.5), combine the acid and base/neutral extracts for each at this point. Do not combine the acid and base/neutral extracts for samples.
10.5 Add a clean boiling chip and attach a two ball micro Snyder column to the concentrator tube. Prewet the column by adding approx 0.5 mL methylene chloride through the top. Place the apparatus in the hot water bath. Adjust the vertical position and the water temperature as required to complete the concentration in 5-10 minutes. At the proper rate of distillation, the balls of the column will actively chatter but the chambers will not flood. When the liquid reaches an apparent volume of approx 0.5 mL, remove the apparatus from the water bath and allow to drain and cool for at least 10 minutes. Remove the micro Snyder column and rinse its lower joint into the concentrator tube with approx 0.2 mL of methylene chloride. Adjust the final volume to 1.0 mL.
10.6 Transfer the concentrated extract to a clean screw-cap vial. Seal the vial with a Teflon-lined lid, and mark the level on the vial. Label with the sample number and fraction, and store in the dark at −20 to −10 °C until ready for analysis.
11. GC/MS Analysis11.1 Establish the operating conditions given in Table 3 or 4 for analysis of the base/neutral or acid extracts, respectively. For analysis of combined extracts (Section 10.4.2), use the operating conditions in Table 3.
11.2 Bring the concentrated extract (Section 10.6) or standard (Sections 6.13 through 6.14) to room temperature and verify that any precipitate has redissolved. Verify the level on the extract (Sections 6.6 and 10.6) and bring to the mark with solvent if required.
11.3 Add the internal standard solution (Section 6.10) to the extract (use 1.0 uL of solution per 0.1 mL of extract) immediately prior to injection to minimize the possibility of loss by evaporation, adsorption, or reaction. Mix thoroughly.
11.4 Inject a volume of the standard solution or extract such that 100 ng of the internal standard will be injected, using on-column or splitless injection. For 1 mL extracts, this volume will be 1.0 uL. Start the GC column initial isothermal hold upon injection. Start MS data collection after the solvent peak elutes. Stop data collection after the benzo (ghi) perylene or pentachlorophenol peak elutes for the base/neutral or acid fraction, respectively. Return the column to the initial temperature for analysis of the next sample.
12. System and Laboratory Performance12.1 At the beginning of each 8 hr shift during which analyses are performed, GC/MS system performance and calibration are verified for all pollutants and labeled compounds. For these tests, analysis of the 100 µg/mL calibration standard (Section 6.13) shall be used to verify all performance criteria. Adjustment and/or recalibration (per Section 7) shall be performed until all performance criteria are met. Only after all performance criteria are met may samples, blanks, and precision and recovery standards be analyzed.
12.2 DFTPP spectrum validity - inject 1 µL of the DFTPP solution (Section 6.11) either separately or within a few seconds of injection of the standard (Section 12.1) analyzed at the beginning of each shift. The criteria in Table 5 shall be met.
12.3 Retention times - the absolute retention time of 2,2′-difluorobiphenyl shall be within the range of 1078 to 1248 seconds and the relative retention times of all pollutants and labeled compounds shall fall within the limits given in Tables 3 and 4.
12.4 GC resolution - the valley height between anthracene and phenanthrene at m/z 178 (or the analogs at m/z 188) shall not exceed 10 percent of the taller of the two peaks.
12.5 Calibration verification - compute the concentration of each pollutant (Tables 1 and 2) by isotope dilution (Section 7.4) for those compounds which have labeled analogs. Compute the concentration of each pollutant which has no labeled analog by the internal standard method (Section 7.5). Compute the concentration of the labeled compounds by the internal standard method. These concentrations are computed based on the calibration data determined in Section 7.
12.5.1 For each pollutant and labeled compound being tested, compare the concentration with the calibration verification limit in Table 8. If all compounds meet the acceptance criteria, calibration has been verified and analysis of blanks, samples, and precision and recovery standards may proceed. If, however, any compound fails, the measurement system is not performing properly for that compound. In this event, prepare a fresh calibration standard or correct the problem causing the failure and repeat the test (Section 12.1), or recalibrate (Section 7).
12.6 Multiple peaks - each compound injected shall give a single, distinct GC peak.
12.7 On-going precision and accuracy.
12.7.1 Analyze the extract of one of the pair of precision and recovery standards (Section 10.1.3) prior to analysis of samples from the same lot.
12.7.2 Compute the concentration of each pollutant (Tables 1 and 2) by isotope dilution (Section 7.4) for those compounds which have labeled analogs. Compute the concentration of each pollutant which has no labeled analog by the internal standard method (Section 7.5). Compute the concentration of the labeled compounds by the internal standard method.
12.7.3 For each pollutant and labeled compound, compare the concentration with the limits for on-going accuracy in Table 8. If all compounds meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may proceed. If, however, any individual concentration falls outside of the range given, system performance is unacceptable for that compound.
Note:The large number of compounds in Table 8 present a substantial probability that one or more will fail when all compounds are analyzed. To determine if the extraction/concentration system is out of control or if the failure is caused by probability, proceed as follows:
12.7.3.1 Analyze the second aliquot of the pair of precision and recovery standard (Section 10.1.3).
12.7.3.2 Compute the concentration of only those pollutants or labeled compounds that failed the previous test (Section 12.7.3). If these compounds now pass, the extraction/concentration processes are in control and analysis of blanks and samples may proceed. If, however, any of the same compounds fail again, the extraction/concentration processes are not being performed properly for these compounds. In this event, correct the problem, re-extract the sample lot (Section 10) and repeat the on-going precision and recovery test (Section 12.7).
12.7.4 Add results which pass the specifications in Section 12.7.2 to initial and previous on-going data. Update QC charts to perform a graphic representation of continued laboratory performance (Figure 5). Develop a statement of laboratory accuracy for each pollutant and labeled compound by calculating the average percent recovery (R) and the standard deviation of percent recovery (sr). Express the accuracy as a recovery interval from R−2sr to R + 2sr. For example, if R = 95% and sr = 5%, the accuracy is 85−105%.
13. Qualitative Determination13.1 Qualititative determination is accomplished by comparison of data from analysis of a sample or blank with data from analysis of the shift standard (Section 12.1) and with data stored in the spectral libraries (Section 7.2.4). Identification is confirmed when spectra and retention times agree per the criteria below.
13.2 Labeled compounds and pollutants having no labeled analog:
13.2.1 The signals for all characteristic masses stored in the spectral library (Section 7.2.4) shall be present and shall maximize within the same two consecutive scans.
13.2.2 Either (1) the background corrected EICP areas, or (2) the corrected relative intensities of the mass spectral peaks at the GC peak maximum shall agree within a factor of two (0.5 to 2 times) for all masses stored in the library.
13.2.3 The retention time relative to the nearest eluted internal standard shall be within ±15 scans or ±15 seconds, whichever is greater of this difference in the shift standard (Section 12.1).
13.3 Pollutants having a labled analog:
13.3.1 The signals for all characteristic masses stored in the spectral library (Section 7.2.4) shall be present and shall maximize within the same two consecutive scans.
13.3.2. Either (1) the background corrected EICP areas, or (2) the corrected relative intensities of the mass spectral peaks at the GC peak maximum shall agree within a factor of two for all masses stored in the spectral library.
13.3.3. The retention time difference between the pollutant and its labeled analog shall agree within ±6 scans or ±6 seconds (whichever is greater) of this difference in the shift standard (Section 12.1).
13.4 Masses present in the experimental mass spectrum that are not present in the reference mass spectrum shall be accounted for by contaminant or background ions. If the experimental mass spectrum is contaminated, an experienced spectrometrist (Section 1.4) is to determine the presence or absence of the cmmpound.
14. Quantitative Determination14.1 Isotope dilution - by adding a known amount of a labeled compound to every sample prior to extraction, correction for recovery of the pollutant can be made because the pollutant and its labeled analog exhibit the same effects upon extraction, concentration, and gas chromatography. Relative response (RR) values for mixtures are used in conjunction with calibration curves described in Section 7.4 to determine concentrations directly, so long as labeled compound spiking levels are constant. For the phenml example given in Figure 1 (Section 7.4.1), RR would be equal to 1.114. For this RR value, the phenol calibration curve given in Figure 1 indicates a concentration of 27 µg/mL in the sample extract (Cex).
14.2 Internal standard - compute the concentration in the extract using the response factor determined from calibration data (Section 7.5) and the following equation: Cex(µg/mL) = (As × Cis/(Ais × RF) where Cex is the concentration of the compound in the extract, and the other terms are as defined in Section 7.5.1.
14.3 The concentration of the pollutant in water is computed using the volumes of the original water sample (Section 10.1) and the final extract volume (Section 10.5), as follows: Concentration in water (µg/L) = (Cex × Vex)/Vs where Vex is the extract volume in mL, and Vs is the sample volume in liters.
14.4 If the EICP area at the quantitiation mass for any compound exceeds the calibration range of the system, the extract of the dilute aliquot (Section 10.1) is analyzed by isotope dilution; otherwise, the extract is diluted by a factor of 10, 9 µL of internal standard solution (Section 6.10) are added to a 1.0 mL aliquot, and this diluted extract is analyzed by the internal standard method (Section 14.2). Quantify each compound at the highest concentration level within the calibration range.
14.5 Report results for all pollutants and labeled compounds (Tables 1 and 2) found in all standards, blanks, and samples in µg/L, to three significant figures. Results for samples which have been diluted are reported at the least dilute level at which the area at the quantitation mass is within the calibration range (Section 14.4) and the labeled compound recovery is within the normal range for the method (Section 15.4).
15. Analysis of Complex Samples15.1 Untreated effluents and other samples frequently contain high levels (>1000 µg/L) of the compounds of interest, interfering compounds, and/or polymeric materials. Some samples will not concentrate to one mL (Section 10.5); others will overload the GC column and/or mass spectrometer.
15.2 Analyze the dilute aliquot (Section 10.1) when the sample will not concentrate to 1.0 mL. If a dilute aliquot was not extracted, and the sample holding time (Section 9.3) has not been exceeded, dilute an aliquot of the sample with reagent water and re-extract (Section 10.1); otherwise, dilute the extract (Section 14.4) and analyze by the internal standard method (Section 14.2).
15.3 Recovery of internal standard - the EICP area of the internal standard should be within a factor of two of the area in the shift standard (Section 12.1). If the absolute areas of the labeled compounds are within a factor of two of the respective areas in the shift standard, and the internal standard area is less than one-half of its respective area, then internal standard loss in the extract has occurred. In this case, use one of the labeled compounds (perferably a polynuclear aromatic hydrocarbon) to compute the concentration of a pollutant with no labeled analog.
15.4 Recovery of labeled compounds - in most samples, labeled compound recoveries will be similar to those from reagent water (Section 12.7). If the labeled compound recovery is outside the limits given in Table 8, the dilute extract (Section 10.1) is analyzed as in Section 14.4. If the recoveries of all labeled compounds and the internal staldard are low (per the criteria above), then a loss in instrument sensitivity is the most likely cause. In this case, the 100 µg/mL calibration standard (Section 12.1) shall be analyzed and calibration verified (Section 12.5). If a loss in sensitivity has occurred, the instrument shall be repaired, the performance specifications in Section 12 shall be met, and the extract reanalyzed. If a loss in instrument sensitivity has not occurred, the method does not work on the sample being analyzed and the result may not be reported for regulatory compliance purposes.
16. Method Performance16.1 Interlaboratory performance for this method is detailed in references 9 and 10.
16.2 A chromatogram of the 100 µg/mL acid/base/neutral calibration standard (Section 6.13) is shown in Figure 6.
References1. “Performance Tests for the Evaluation of Computerized Gas Chromatography/Mass Spectrometry Equipment and Laboratories” USEPA, EMSL/Cincinnati, OH 45268, EPA-600/4-80-025 (April 1980).
2. “Working with Carcinogens,” DHEW, PHS, CDC, NIOSH, Publication 77-206, (August 1977).
3. “OSHA Safety and Health Standards, General Industry” OSHA 2206, 29 CFR part 1910 (January 1976).
4. “Safety in Academic Chemistry Laboratories, ” ACS Committee on Chemical Safety (1979).
5. “Reference Compound to Calibrate Ion Abundance Measurement in Gas Chromatography-Mass Spectrometry Systems,” J.W. Eichelberger, L.E. Harris, and W.L. Budde, Anal. Chem., 47, 955 (1975).
6. “Handbook of Analytical Quality Control in Water and Wastewater Laboratories,” USEPA, EMSL/Cincinnati, OH 45268, EPA-600/4-79-019 (March 1979).
7. “Standard Practice for Sampling Water,” ASTM Annual Book of Standards, ASTM, Philadelphia, PA, 76 (1980).
8. “Methods 330.4 and 330.5 for Total Residual Chlorine,” USEPA, EMSL/ Cincinnati, OH 45268, EPA 600/4-70-020 (March 1979).
9. Colby, B.N., Beimer, R.G., Rushneck, D.R., and Telliard, W.A., “Isotope Dilution Gas Chromatography-Mass Spectrometry for the determination of Priority Pollutants in Industrial Effluents.” USEPA, Effluent Guidelines Division, Washington, DC 20460 (1980).
10. “Inter-laboratory Validation of US Environmental Protection Agency Method 1625,” USEPA, Effluent Guidelines Division, Washington, DC 20460 (June 15, 1984).
Table 1 - Base/Neutral Extractable Compounds
| Compound | STORET | CAS registry | EPA-EGD | NPDES |
|---|---|---|---|---|
| Acenaphthene | 34205 | 83-32-9 | 001 B | 001 B |
| Acenaphthylene | 34200 | 208-96-8 | 077 B | 002 B |
| Anthracene | 34220 | 120-12-7 | 078 B | 003 B |
| Benzidine | 39120 | 92-87-5 | 005 B | 004 B |
| Benzo(a)anthracene | 34526 | 56-55-3 | 072 B | 005 B |
| Benzo(b)fluoranthene | 34230 | 205-99-2 | 074 B | 007 B |
| Benzo(k)fluoranthene | 34242 | 207-08-9 | 075 B | 009 B |
| Benzo(a)pyrene | 34247 | 50-32-8 | 073 B | 006 B |
| Benzo(ghi)perylene | 34521 | 191-24-2 | 079 B | 008 B |
| Biphenyl (Appendix C) | 81513 | 92-52-4 | 512 B | |
| Bis(2-chloroethyl) ether | 34273 | 111-44-4 | 018 B | 011 B |
| Bis(2-chloroethyoxy)methane | 34278 | 111-91-1 | 043 B | 010 B |
| Bis(2-chloroisopropyl) ether | 34283 | 108-60-1 | 042 B | 012 B |
| Bis(2-ethylhexyl) phthalate | 39100 | 117-81-7 | 066 B | 013 B |
| 4-bromophenyl phenyl ether | 34636 | 101-55-3 | 041 B | 014 B |
| Butyl benzyl phthalate | 34292 | 85-68-7 | 067 B | 015 B |
| n-C10 (Appendix C) | 77427 | 124-18-5 | 517 B | |
| n-C12 (Appendix C) | 77588 | 112-40-2 | 506 B | |
| n-C14 (Appendix C) | 77691 | 629-59-4 | 518 B | |
| n-C16 (Appendix C) | 77757 | 544-76-3 | 519 B | |
| n-C18 (Appendix C) | 77804 | 593-45-3 | 520 B | |
| n-C20 (Appendix C) | 77830 | 112-95-8 | 521 B | |
| n-C22 (Appendix C) | 77859 | 629-97-0 | 522 B | |
| n-C24 (Appendix C) | 77886 | 646-31-1 | 523 B | |
| n-C26 (Appendix C) | 77901 | 630-01-3 | 524 B | |
| n-C28 (Appendix C) | 78116 | 630-02-4 | 525 B | |
| n-C30 (Appendix C) | 78117 | 638-68-6 | 526 B | |
| Carbazole (4c) | 77571 | 86-74-8 | 528 B | |
| 2-chloronaphthalene | 34581 | 91-58-7 | 020 B | 016 B |
| 4-chlorophenyl phenyl ether | 34641 | 7005-72-3 | 040 B | 017 B |
| Chrysene | 34320 | 218-01-9 | 076 B | 018 B |
| P-cymene (Appendix C) | 77356 | 99-87-6 | 513 B | |
| Dibenzo(a,h)anthracene | 34556 | 53-70-3 | 082 B | 019 B |
| Dibenzofuran (Appendix C and 4c) | 81302 | 132-64-9 | 505 B | |
| Dibenzothiophene (Synfuel) | 77639 | 132-65-0 | 504 B | |
| Di-n-butyl phthalate | 39110 | 84-74-2 | 068 B | 026 B |
| 1,2-dichlorobenzene | 34536 | 95-50-1 | 025 B | 020 B |
| 1,3-dichlorobenzene | 34566 | 541-73-1 | 026 B | 021 B |
| 1,4-dichlorobenzene | 34571 | 106-46-7 | 027 B | 022 B |
| 3,3′-dichlorobenzidine | 34631 | 91-94-1 | 028 B | 023 B |
| Diethyl phthalate | 34336 | 84-66-2 | 070 B | 024 B |
| 2,4-dimethylphenol | 34606 | 105-67-9 | 034 A | 003 A |
| Dimethyl phthalate | 34341 | 131-11-3 | 071 B | 025 B |
| 2,4-dinitrotoluene | 34611 | 121-14-2 | 035 B | 027 B |
| 2,6-dinitrotoluene | 34626 | 606-20-2 | 036 B | 028 B |
| Di-n-octyl phthalate | 34596 | 117-84-0 | 069 B | 029 B |
| Diphenylamine (Appendix C) | 77579 | 122-39-4 | 507 B | |
| Diphenyl ether (Appendix C) | 77587 | 101-84-8 | 508 B | |
| 1,2-diphenylhydrazine | 34346 | 122-66-7 | 037 B | 030 B |
| Fluoranthene | 34376 | 206-44-0 | 039 B | 031 B |
| Fluorene | 34381 | 86-73-7 | 080 B | 032 B |
| Hexachlorobenzene | 39700 | 118-74-1 | 009 B | 033 B |
| Hexachlorobutadiene | 34391 | 87-68-3 | 052 B | 034 B |
| Hexachloroethane | 34396 | 67-72-1 | 012 B | 036 B |
| Hexachlorocyclopentadiene | 34386 | 77-47-4 | 053 B | 035 B |
| Indeno(1,2,3-cd)pyrene | 34403 | 193-39-5 | 083 B | 037 B |
| Isophorone | 34408 | 78-59-1 | 054 B | 038 B |
| Naphthalene | 34696 | 91-20-3 | 055 B | 039 B |
| B-naphthylamine (Appendix C) | 82553 | 91-59-8 | 502 B | |
| Nitrobenzene | 34447 | 98-95-3 | 056 B | 040 B |
| N-nitrosodimethylamine | 34438 | 62-75-9 | 061 B | 041 B |
| N-nitrosodi-n-propylamine | 34428 | 621-64-7 | 063 B | 042 B |
| N-nitrosodiphenylamine | 34433 | 86-30-3 | 062 B | 043 B |
| Phenanthrene | 34461 | 85-01-8 | 081 B | 044 B |
| Phenol | 34694 | 108-95-2 | 065 A | 010 A |
| a-Picoline (Synfuel) | 77088 | 109-06-89 | 503 B | |
| Pyrene | 34469 | 129-00-0 | 084 B | 045 B |
| styrene (Appendix C) | 77128 | 100-42-5 | 510 B | |
| a-terpineol (Appendix C) | 77493 | 98-55-5 | 509 B | |
| 1,2,3-trichlorobenzene (4c) | 77613 | 87-61-6 | 529 B | |
| 1,2,4-trichlorobenzene | 34551 | 120-82-1 | 008 B | 046 B |
Table 2 - Acid Extractable Compounds
| Compound | STORET | CAS registry | EPA-EGD | NPDES |
|---|---|---|---|---|
| 4-chloro-3-methylphenol | 34452 | 59-50-7 | 022 A | 008 A |
| 2-chlorophenol | 34586 | 95-57-8 | 024 A | 001 A |
| 2,4-dichlorophenol | 34601 | 120-83-2 | 031 A | 002 A |
| 2,4-dinitrophenol | 34616 | 51-28-5 | 059 A | 005 A |
| 2-methyl-4,6-dinitrophenol | 34657 | 534-52-1 | 060 A | 004 A |
| 2-nitrophenol | 34591 | 88-75-5 | 057 A | 006 A |
| 4-nitrophenol | 34646 | 100-02-7 | 058 A | 007 A |
| Pentachlorophenol | 39032 | 87-86-5 | 064 A | 009 A |
| 2,3,6-trichlorophenol (4c) | 77688 | 93-37-55 | 530 A | |
| 2,4,5-trichlorophenol (4c) | 95-95-4 | 531 A | ||
| 2,4,6-trichlorophenol | 34621 | 88-06-2 | 021 A | 011 A |
Table 3 - Gas Chromatography of Base/Neutral Extractable Compounds
| EGD No. 1 | Compound | Retention time | Detection limit 2 (µg/L) | ||
|---|---|---|---|---|---|
| Mean (sec) | EGD Ref | Relative | |||
| 164 | 2,2′-difluorobiphenyl (int std) | 1163 | 164 | 1.000-1.000 | 10 |
| 061 | N-nitrosodimethylamine | 385 | 164 | ns | 50 |
| 603 | alpha picoline-d7 | 417 | 164 | 0.326-0.393 | 50 |
| 703 | alpha picoline | 426 | 603 | 1.006-1.028 | 50 |
| 610 | styrene-d5 | 546 | 164 | 0.450-0.488 | 10 |
| 710 | styrene | 549 | 610 | 1.002-1.009 | 10 |
| 613 | p-cymene-d14 | 742 | 164 | 0.624-0.652 | 10 |
| 713 | p-cymene | 755 | 613 | 1.008-1.023 | 10 |
| 265 | phenol-d5 | 696 | 164 | 0.584-0.613 | 10 |
| 365 | phenol | 700 | 265 | 0.995-1.010 | 10 |
| 218 | bis(2-chloroethyl) ether-d8 | 696 | 164 | 0.584-0.607 | 10 |
| 318 | bis(2-chloroethyl) ether | 704 | 218 | 1.007-1.016 | 10 |
| 617 | n-decane-d22 | 698 | 164 | 0.585-0.615 | 10 |
| 717 | n-decane | 720 | 617 | 1.022-1.038 | 10 |
| 226 | 1,3-dichlorobenzene-d4 | 722 | 164 | 0.605-0.636 | 10 |
| 326 | 1,3-dichlorobenzene | 724 | 226 | 0.998-1.008 | 10 |
| 227 | 1,4-dichlorobenzene-d4 | 737 | 164 | 0.601-0.666 | 10 |
| 327 | 1,4-dichlorobenzene | 740 | 227 | 0.997-1.009 | 10 |
| 225 | 1,2-dichlorobenzene-d4 | 758 | 164 | 0.632-0.667 | 10 |
| 325 | 1,2-dichlorobenzene | 760 | 225 | 0.995-1.008 | 10 |
| 242 | bis(2-chloroisopropyl) ether-d12 | 788 | 164 | 0.664-0.691 | 10 |
| 342 | bis(2-chloroisopropyl) ether | 799 | 242 | 1.010-1.016 | 10 |
| 212 | hexachloroethane-13C | 819 | 164 | 0.690-0.717 | 10 |
| 312 | hexachloroethane | 823 | 212 | 0.999-1.001 | 10 |
| 063 | N-nitrosodi-n-propylamine | 830 | 164 | ns | 20 |
| 256 | nitrobenzene-d5 | 845 | 164 | 0.706-0.727 | 10 |
| 356 | nitrobenzene | 849 | 256 | 1.002-1.007 | 10 |
| 254 | isophorone-d8 | 881 | 164 | 0.747-0.767 | 10 |
| 354 | isophorone | 889 | 254 | 0.999-1.017 | 10 |
| 234 | 2,4-dimethyl phenol-d3 | 921 | 164 | 0.781-0.803 | 10 |
| 334 | 2,4-dimethylphenol | 924 | 234 | 0.999-1.003 | 10 |
| 043 | bis(2-chloroethoxy) methane | 939 | 164 | ns | 10 |
| 208 | 1,2,4-trichlorobenzene-d3 | 955 | 164 | 0.813-0.830 | 10 |
| 308 | 1,2,4-trichlorobenzene | 958 | 208 | 1.000-1.005 | 10 |
| 255 | naphthalene-d8 | 963 | 164 | 0.819-0.836 | 10 |
| 355 | naphthalene | 967 | 255 | 1.001-1.006 | 10 |
| 609 | alpha-terpineol-d3 | 973 | 164 | 0.829-0.844 | 10 |
| 709 | alpha-terpineol | 975 | 609 | 0.998-1.008 | 10 |
| 606 | n-dodecane-d26 | 953 | 164 | 0.730-0.908 | 10 |
| 706 | n-dodecane | 981 | 606 | 0.986-1.051 | 10 |
| 529 | 1,2,3-trichlorobenzene | 1003 | 164 | ns | 10 |
| 252 | hexachlorobutadiene-13C4 | 1005 | 164 | 0.856-0.871 | 10 |
| 352 | hexachlorobutadiene | 1006 | 252 | 0.999-1.002 | 10 |
| 253 | hexachlorocyclopentadiene-13C4 | 1147 | 164 | 0.976-0.986 | 10 |
| 353 | hexachlorocyclopentadiene | 1142 | 253 | 0.999-1.001 | 10 |
| 220 | 2-chloronaphthalene-d7 | 1185 | 164 | 1.014-1.024 | 10 |
| 320 | 2-chloronaphthalene | 1200 | 220 | 0.997-1.007 | 10 |
| 518 | n-tetradecane | 1203 | 164 | ns | 10 |
| 612 | Biphenyl-d10 | 1205 | 164 | 1.016-1.027 | 10 |
| 712 | Biphenyl | 1195 | 612 | 1.001-1.006 | 10 |
| 608 | Diphenyl ether-d10 | 1211 | 164 | 1.036-1.047 | 10 |
| 708 | Diphenyl ether | 1216 | 608 | 0.997-1.009 | 10 |
| 277 | Acenaphthylene-d8 | 1265 | 164 | 1.080-1.095 | 10 |
| 377 | Acenaphthylene | 1247 | 277 | 1.000-1.004 | 10 |
| 271 | Dimethyl phthalate-d4 | 1269 | 164 | 1.083-1.102 | 10 |
| 371 | Dimethyl phthalate | 1273 | 271 | 0.998-1.005 | 10 |
| 236 | 2,6-dinitrotoluene-d3 | 1283 | 164 | 1.090-1.112 | 10 |
| 336 | 2,6-dinitrotoluene | 1300 | 236 | 1.001-1.005 | 10 |
| 201 | Acenaphthene-d10 | 1298 | 164 | 1.107-1.125 | 10 |
| 301 | Acenaphthene | 1304 | 201 | 0.999-1.009 | 10 |
| 605 | Dibenzofuran-d8 | 1331 | 164 | 1.134-1.155 | 10 |
| 705 | Dibenzofuran | 1335 | 605 | 0.998-1.007 | 10 |
| 602 | Beta-naphthylamine-d7 | 1368 | 164 | 1.163-1.189 | 50 |
| 702 | Beta-naphthylamine | 1371 | 602 | 0.996-1.007 | 50 |
| 280 | Fluorene-d10 | 1395 | 164 | 1.185-1.214 | 10 |
| 380 | Fluorene | 1401 | 281 | 0.999-1.008 | 10 |
| 240 | 4-chlorophenyl phenyl ether-d5 | 1406 | 164 | 1.194-1.223 | 10 |
| 340 | 4-chlorophenyl phenyl ether | 1409 | 240 | 0.990-1.015 | 10 |
| 270 | Diethyl phthalate-d4 | 1409 | 164 | 1.197-1.229 | 10 |
| 370 | Diethyl phthalate | 1414 | 270 | 0.996-1.006 | 10 |
| 619 | n-hexadecane-d34 | 1447 | 164 | 1.010-1.478 | 10 |
| 719 | n-hexadecane | 1469 | 619 | 1.013-1.020 | 10 |
| 235 | 2,4-dinitrotoluene-d3 | 1359 | 164 | 1.152-1.181 | 10 |
| 335 | 2,4-dinitrotoluene | 1344 | 235 | 1.000-1.002 | 10 |
| 237 | 1,2-diphenylhydrazine-d8 | 1433 | 164 | 1.216-1.248 | 20 |
| 337 | 1,2-diphenylhydrazine ( 3) | 1439 | 237 | 0.999-1.009 | 20 |
| 607 | Diphenylamine-d10 | 1437 | 164 | 1.213-1.249 | 20 |
| 707 | Diphenylamine | 1439 | 607 | 1.000-1.007 | 20 |
| 262 | N-nitrosodiphenylamine-d6 | 1447 | 164 | 1.225-1.252 | 20 |
| 362 | N-nitrosodiphenylamine ( 4) | 1464 | 262 | 1.000-1.002 | 20 |
| 041 | 4-bromophenyl phenyl ether | 1498 | 164 | 1.271-1.307 | 10 |
| 209 | Hexachlorobenzene-13C6 | 1521 | 164 | 1.288-1.327 | 10 |
| 309 | Hexachlorobenzene | 1522 | 209 | 0.999-1.001 | 10 |
| 281 | Phenanthrene-d10 | 1578 | 164 | 1.334-1.380 | 10 |
| 520 | n-octadecane | 1580 | 164 | ns | 10 |
| 381 | Phenanthrene | 1583 | 281 | 1.000-1.005 | 10 |
| 278 | Anthracene-d10 | 1588 | 164 | 1.342-1.388 | 10 |
| 378 | Anthracene | 1592 | 278 | 0.998-1.006 | 10 |
| 604 | Dibenzothiophene-d8 | 1559 | 164 | 1.314-1.361 | 10 |
| 704 | Dibenzothiophene | 1564 | 604 | 1.000-1.006 | 10 |
| 528 | Carbazole | 1650 | 164 | ns | 20 |
| 621 | n-eicosane-d42 | 1655 | 164 | 1.184-1.662 | 10 |
| 721 | n-eicosane | 1677 | 621 | 1.010-1.021 | 10 |
| 268 | Di-n-butyl phthalate-d4 | 1719 | 164 | 1.446-1.510 | 10 |
| 368 | Di-n-butyl phthalate | 1723 | 268 | 1.000-1.003 | 10 |
| 239 | Fluoranthene-d10 | 1813 | 164 | 1.522-1.596 | 10 |
| 339 | Fluoranthene | 1817 | 239 | 1.000-1.004 | 10 |
| 284 | Pyrene-d10 | 1844 | 164 | 1.523-1.644 | 10 |
| 384 | Pyrene | 1852 | 284 | 1.001-1.003 | 10 |
| 205 | Benzidine-d8 | 1854 | 164 | 1.549-1.632 | 50 |
| 305 | Benzidine | 1853 | 205 | 1.000-1.002 | 50 |
| 522 | n-docosane | 1889 | 164 | ns | 10 |
| 623 | n-tetracosane-d50 | 1997 | 164 | 1.671-1.764 | 10 |
| 723 | n-tetracosane | 2025 | 612 | 1.012-1.015 | 10 |
| 067 | Butylbenzyl phthalate | 2060 | 164 | ns | 10 |
| 276 | Chrysene-d12 | 2081 | 164 | 1.743-1.837 | 10 |
| 376 | Chrysene | 2083 | 276 | 1.000-1.004 | 10 |
| 272 | Benzo(a)anthracene-d12 | 2082 | 164 | 1.735-1.846 | 10 |
| 372 | Benzo(a)anthracene | 2090 | 272 | 0.999-1.007 | 10 |
| 228 | 3,3′-dichlorobenzidine-d6 | 2088 | 164 | 1.744-1.848 | 50 |
| 328 | 3,3′-dichlorobenzidine | 2086 | 228 | 1.000-1.001 | 50 |
| 266 | Bis(2-ethylhexyl) phthalate-d4 | 2123 | 164 | 1.771-1.880 | 10 |
| 366 | Bis(2-ethylhexyl) phthalate | 2124 | 266 | 1.000-1.002 | 10 |
| 524 | n-hexacosane | 2147 | 164 | ns | 10 |
| 269 | di-n-octyl phthalate-d4 | 2239 | 164 | 1.867-1.982 | 10 |
| 369 | di-n-octyl phthalate | 2240 | 269 | 1.000-1.002 | 10 |
| 525 | n-octacosane | 2272 | 164 | ns | 10 |
| 274 | Benzo(b)fluoranthene-d12 | 2281 | 164 | 1.902-2.025 | 10 |
| 354 | Benzo(b)fluoranthene | 2293 | 274 | 1.000-1.005 | 10 |
| 275 | Benzo(k)fluoranthene-d12 | 2287 | 164 | 1.906-2.033 | 10 |
| 375 | Benzo(k)fluoranthene | 2293 | 275 | 1.000-1.005 | 10 |
| 273 | Benzo(a)pyrene-d12 | 2351 | 164 | 1.954-2.088 | 10 |
| 373 | Benzo(a)pyrene | 2350 | 273 | 1.000-1.004 | 10 |
| 626 | N-triacontane-d62 | 2384 | 164 | 1.972-2.127 | 10 |
| 726 | N-triacontane | 2429 | 626 | 1.011-1.028 | 10 |
| 083 | Indeno(1,2,3-cd)pyrene | 2650 | 164 | ns | 20 |
| 082 | Dibenzo(a,h)anthracene | 2660 | 164 | ns | 20 |
| 279 | Benzo(ghi)perylene-d12 | 2741 | 164 | 2.187-2.524 | 20 |
| 379 | Benzo(ghi)perylene | 2750 | 279 | 1.001-1.006 | 20 |
1 Reference numbers beginning with 0, 1 or 5 indicate a pollutant quantified by the internal standard method; reference numbers beginning with 2 or 6 indicate a labeled compound quantified by the internal standard method; reference numbers beginning with 3 or 7 indicate a pollutant quantified by isotope dilution.
2 This is a minimum level at which the entire GC/MS system must give recognizable mass spectra (background corrected) and acceptable calibration points.
3 Detected as azobenzene.
4 Detected as diphenylamine.
ns = specification not available at time of release of method.
Column: 30 ±2 m × 0.25 ±0.02 mm i.d. 94% methyl, 4% phenyl, 1% vinyl bonded phase fused silica capillary.
Temperature program: 5 min at 30 °C; 30 - 280 °C at 8 °C per min; isothermal at 280 °C until benzo(ghi)perylene elutes.
Gas velocity: 30 ±5 cm/sec.
Table 4 - Gas Chromatography of Acid Extractable Compounds
| EGD No. 1 | Compound | Retention time | Detection limit 2 (µg/L) | ||
|---|---|---|---|---|---|
| Mean (sec) | EGD Ref | Relative | |||
| 164 | 2,2′-difluorobiphenyl (int std) | 1163 | 164 | 1.000-1.000 | 10 |
| 224 | 2-chlorophenol-d4 | 701 | 164 | 0.587-0.618 | 10 |
| 324 | 2-chlorophenol | 705 | 224 | 0.997-1.010 | 10 |
| 257 | 2-nitrophenol-d4 | 898 | 164 | 0.761-0.783 | 20 |
| 357 | 2-nitrophenol | 900 | 257 | 0.994-1.009 | 20 |
| 231 | 2,4-dichlorophenol-d3 | 944 | 164 | 0.802-0.822 | 10 |
| 331 | 2,4-dichlorophenol | 947 | 231 | 0.997-1.006 | 10 |
| 222 | 4-chloro-3-methylphenol-d2 | 1086 | 164 | 0.930-0.943 | 10 |
| 322 | 4-chloro-3-methylphenol | 1091 | 222 | 0.998-1.003 | 10 |
| 221 | 2,4,6-trichlorophenol-d2 | 1162 | 164 | 0.994-1.005 | 10 |
| 321 | 2,4,6-trichlorophenol | 1165 | 221 | 0.998-1.004 | 10 |
| 531 | 2,4,5-trichlorophenol | 1170 | 164 | ns | 10 |
| 530 | 2,3,6-trichlorophenol | 1195 | 164 | ns | 10 |
| 259 | 2,4-dinitrophenol-d3 | 1323 | 164 | 1.127-1.149 | 50 |
| 359 | 2,4-dinitrophenol | 1325 | 259 | 1.000-1.005 | 50 |
| 258 | 4-nitrophenol-d4 | 1349 | 164 | 1.147-1.175 | 50 |
| 358 | 4-nitrophenol | 1354 | 258 | 0.997-1.006 | 50 |
| 260 | 2-methyl-4,6-dinitrophenol-d2 | 1433 | 164 | 1.216-1.249 | 20 |
| 360 | 2-methyl-4,6-dinitrophenol | 1435 | 260 | 1.000-1.002 | 20 |
| 264 | Pentachlorophenol-13C6 | 1559 | 164 | 1.320-1.363 | 50 |
| 364 | Pentachlorophenol | 1561 | 264 | 0.998-1.002 | 50 |
1 Reference numbers beginning with 0, 1 or 5 indicate a pollutant quantified by the internal standard method; reference numbers beginning with 2 or 6 indicate a labeled compound quantified by the internal standard method; reference numbers beginning with 3 or 7 indicate a pollutant quantified by isotope dilution.
2 This is a minimum level at which the entire GC/MS system must give recognizable mass spectra (background corrected) and acceptable calibration points.
ns = specification not available at time of release of method.
Column: 30 ±2m × 0.25 ±0.02mm i.d. 94% methyl, 4% phenyl, 1% vinyl bonded phase fused silica capillary.
Temperature program: 5 min at 30 °C; 8 °C/min. to 250 °C or until pentachlorophenol elutes.
Gas velocity: 30 ±5 cm/sec.
Table 5 - DFTPP Mass Intensity Specifications
| Mass | Intensity required |
|---|---|
| 51 | 30-60 percent of mass 198. |
| 68 | Less than 2 percent of mass 69. |
| 70 | Less than 2 percent of mass 69. |
| 127 | 40-60 percent of mass 198. |
| 197 | Less than 1 percent of mass 198. |
| 199 | 5-9 percent of mass 198. |
| 275 | 10-30 percent of mass 198. |
| 365 | greater than 1 percent of mass 198 |
| 441 | present and less than mass 443 |
| 442 | 40-100 percent of mass 198. |
| 443 | 17-23 percent of mass 442. |
Table 6 - Base/Neutral Extractable Compound Characteristic Masses
| Compound | Labeled analog | Primary m/z |
|---|---|---|
| Acenaphthene | d10 | 154/164 |
| Acenaphthylene | d8 | 152/160 |
| Anthracene | d10 | 178/188 |
| Benzidine | d8 | 184/192 |
| Benzo(a)anthracene | d12 | 228/240 |
| Benzo(b)fluoranthene | d12 | 252/264 |
| Benzo(k)fluoranthene | d12 | 252/264 |
| Benzo(a)pyrene | d12 | 252/264 |
| Benzo(ghi)perylene | d12 | 276/288 |
| Biphenyl | d10 | 154/164 |
| Bis(2-chloroethyl) ether | d8 | 93/101 |
| Bis(2-chloroethoxy)methane | 93 | |
| Bis(2-chloroisopropyl) ether | d12 | 121/131 |
| Bis(2-ethylhexyl) phthalate | d4 | 149/153 |
| 4-bromophenyl phenyl ether | 248 | |
| Butyl benzyl phthalate | 149 | |
| n-C10 | d22 | 55/66 |
| n-C12 | d26 | 55/66 |
| n-C14 | 55 | |
| n-C16 | d34 | 55/66 |
| n-C18 | 55 | |
| n-C20 | d42 | 55/66 |
| n-C22 | 55 | |
| n-C24 | d50 | 55/66 |
| n-C26 | 55 | |
| n-C28 | 55 | |
| n-C30 | d62 | 55/66 |
| Carbazole | d8 | 167/175 |
| 2-chloronaphthalene | d7 | 162/169 |
| 4-chlorophenyl phenyl ether | d5 | 204/209 |
| Chrysene | d12 | 228/240 |
| p-cymene | d14 | 114/130 |
| Dibenzo(a,h)anthracene | 278 | |
| Dibenzofuran | d8 | 168/176 |
| Dibenzothiophene | d8 | 184/192 |
| Di-n-butyl phthalate | d4 | 149/153 |
| 1,2-dichlorobenzene | d4 | 146/152 |
| 1,3-dichlorobenzene | d4 | 146/152 |
| 1,4-dichlorobenzene | d4 | 146/152 |
| 3,3′-dichlorobenzidine | d6 | 252/258 |
| Diethyl phthalate | d4 | 149/153 |
| 2,4-dimethylphenol | d3 | 122/125 |
| Dimethyl phthalate | d4 | 163/167 |
| 2,4-dinitrotoluene | d3 | 164/168 |
| 2,6-dinitrotoluene | d3 | 165/167 |
| Di-n-octyl phthalate | d4 | 149/153 |
| Diphenylamine | d10 | 169/179 |
| Diphenyl ether | d10 | 170/180 |
| 1,2-diphenylhydrazine 1 | d10 | 77/82 |
| Fluoranthene | d10 | 202/212 |
| Fluorene | d10 | 166/176 |
| Hexachlorobenzene | 13C6 | 284/292 |
| Hexachlorobutadiene | 13C4 | 225/231 |
| Hexachloroethane | 13C | 201/204 |
| Hexachlorocyclopentadiene | 13C4 | 237/241 |
| Ideno(1,2,3-cd)pyrene | 276 | |
| Isophorone | d8 | 82/88 |
| Naphthalene | d8 | 128/136 |
| B-naphthylamine | d7 | 143/150 |
| Nitrobenzene | d5 | 123/128 |
| N-nitrosodimethylamine | 74 | |
| N-nitrosodi-n-propylamine | 70 | |
| N-nitrosodiphenylamile 2 | d6 | 169/175 |
| Phenanthrene | d10 | 178/188 |
| Phenol | d5 | 94/71 |
| a-picoline | d7 | 93/100 |
| Pyrene | d10 | 202/212 |
| Styrene | d5 | 104/109 |
| a-terpineol | d3 | 59/62 |
| 1,2,3-trichlorobenzene | d3 | 180/183 |
| 1,2,4-trichlorobenzene | d3 | 180/183 |
1 Detected as azobenzene.
2 Detected as diphenylamine.
Table 7 - Acid Extractable Compound Characteristic Masses
| Compound | Labeled analog | Primary m/z |
|---|---|---|
| 4-chloro-3-methylphenol | d2 | 107/109 |
| 2-chlorophenol | d4 | 128/132 |
| 2,4-dichlorophenol | d3 | 162/167 |
| 2,4-dinitrophenol | d3 | 184/187 |
| 2-methyl-4,6-dinitrophenol | d2 | 198/200 |
| 2-nitrophenol | d4 | 139/143 |
| 4-nitrophenol | d4 | 139/143 |
| Pentachlorophenol | 13C6 | 266/272 |
| 2,3,6-trichlorophenol | d2 | 196/200 |
| 2,4,5-trichlorophenol | d2 | 196/200 |
| 2,4,6-trichlorophenol | d2 | 196/200 |
Table 8 - Acceptance Criteria for Performance Tests
| EGD No. 1 | Compound | Acceptance criteria | ||||
|---|---|---|---|---|---|---|
| Initial precision and accuracy section 8.2.3 (µg/L) | Labeled compound recovery sec. 8.3 and 14.2 P (percent) | Calibration verification sec. 12.5 (µg/mL) | On-going accuracy sec. 11.6 R (µg/L) | |||
| s | X | |||||
| 301 | Acenaphthene | 21 | 79-134 | 80-125 | 72-144 | |
| 201 | Acenaphthene-d10 | 38 | 38-147 | 20-270 | 71-141 | 30-180 |
| 377 | Acenaphtylene | 38 | 69-186 | 60-166 | 61-207 | |
| 277 | Acenaphthylene-d8 | 31 | 38-146 | 23-239 | 66-152 | 33-168 |
| 378 | Anthracene | 41 | 58-174 | 60-168 | 50-199 | |
| 278 | Anthracene-d10 | 49 | 31-194 | 14-419 | 58-171 | 23-242 |
| 305 | Benzidine | 119 | 16-518 | 34-296 | 11-672 | |
| 205 | Benzidine-d8 | 269 | ns-ns | ns-ns | ns-ns | ns-ns |
| 372 | Benzo(a)anthracene | 20 | 65-168 | 70-142 | 62-176 | |
| 272 | Benzo(a)anthracene-d12 | 41 | 25-298 | 12-605 | 28-357 | 22-329 |
| 374 | Benzo(b)fluoranthene | 183 | 32-545 | 61-164 | 20-ns | |
| 274 | Benzo(b)fluoranthene-d12 | 168 | 11-577 | ns-ns | 14-ns | ns-ns |
| 375 | Benzo(k)fluoranthene | 26 | 59-143 | 13-ns | 53-155 | |
| 275 | Benzo(k)fluoranthene-d12 | 114 | 15-514 | ns-ns | 13-ns | ns-685 |
| 373 | Benzo(a)pyrene | 26 | 62-195 | 78-129 | 59-206 | |
| 273 | Benzo(a)pyrene-d12 | 24 | 35-181 | 21-290 | 12-ns | 32-194 |
| 379 | Benzo(ghi)perylene | 21 | 72-160 | 69-145 | 58-168 | |
| 279 | Benzo(ghi)perylene-d12 | 45 | 29-268 | 14-529 | 13-ns | 25-303 |
| 712 | Biphenyl (Appendix C) | 41 | 75-148 | 58-171 | 62-176 | |
| 612 | Biphenyl-d12 | 43 | 28-165 | ns-ns | 52-192 | 17-267 |
| 318 | Bis(2-chloroethyl) ether | 34 | 55-196 | 61-164 | 50-213 | |
| 218 | Bis(2-chloroethyl) ether-d8 | 33 | 29-196 | 15-372 | 52-194 | 25-222 |
| 043 | Bis(2-chloroethoxy)methane* | 27 | 43-153 | 44-228 | 39-166 | |
| 342 | Bis(2-chloroisopropyl) ether | 17 | 81-138 | 67-148 | 77-145 | |
| 242 | Bis(2-chloroisopropyl)ether-d12 | 27 | 35-149 | 20-260 | 44-229 | 30-169 |
| 366 | Bis(2-ethylhexyl) phthalate | 31 | 69-220 | 76-131 | 64-232 | |
| 266 | Bis(2-ethylhexyl) phthalate-d4 | 29 | 32-205 | 18-364 | 43-232 | 28-224 |
| 041 | 4-bromophenyl phenyl ether* | 44 | 44-140 | 52-193 | 35-172 | |
| 067 | Butyl benzyl phthalate* | 31 | 19-233 | 22-450 | 35-170 | |
| 717 | n-C10 (Appendix C) | 51 | 24-195 | 42-235 | 19-237 | |
| 617 | n-C10-d22 | 70 | ns-298 | ns-ns | 44-227 | ns-504 |
| 706 | n-C12 (Appendix C) | 74 | 35-369 | 60-166 | 29-424 | |
| 606 | n-C12-d26 | 53 | ns-331 | ns-ns | 41-242 | ns-408 |
| 518 | n-C14 (Appendix C)* | 109 | ns-985 | 37-268 | ns-ns | |
| 719 | n-C16 (Appendix C) | 33 | 80-162 | 72-138 | 71-181 | |
| 619 | n-C16-d34 | 46 | 37-162 | 18-308 | 54-186 | 28-202 |
| 520 | n-C18 (Appendix C)* | 39 | 42-131 | 40-249 | 35-167 | |
| 721 | n-C20 (Appendix C) | 59 | 53-263 | 54-184 | 46-301 | |
| 621 | n-C20-d42 | 34 | 34-172 | 19-306 | 62-162 | 29-198 |
| 522 | n-C22 (Appendix C)* | 31 | 45-152 | 40-249 | 39-195 | |
| 723 | n-C24 (Appendix C) | 11 | 80-139 | 65-154 | 78-142 | |
| 623 | n-C24-d50 | 28 | 27-211 | 15-376 | 50-199 | 25-229 |
| 524 | n-C26 (Appendix C)* | 35 | 35-193 | 26-392 | 31-212 | |
| 525 | n-C28 (Appendix C)* | 35 | 35-193 | 26-392 | 31-212 | |
| 726 | n-C30 (Appendix C) | 32 | 61-200 | 66-152 | 56-215 | |
| 626 | n-C30-d62 | 41 | 27-242 | 13-479 | 24-423 | 23-274 |
| 528 | Carbazole (4c)* | 38 | 36-165 | 44-227 | 31-188 | |
| 320 | 2-chloronaphthalene | 100 | 46-357 | 58-171 | 35-442 | |
| 220 | 2-chloronaphthalene-d7 | 41 | 30-168 | 15-324 | 72-139 | 24-204 |
| 322 | 4-chloro-3-methylphenol | 37 | 76-131 | 85-115 | 62-159 | |
| 222 | 4-chloro-3-methylphenol-d2 | 111 | 30-174 | ns-613 | 68-147 | 14-314 |
| 324 | 2-chlorophenol | 13 | 79-135 | 78-129 | 76-138 | |
| 224 | 2-chlorophenol-d4 | 24 | 36-162 | 23-255 | 55-180 | 33-176 |
| 340 | 4-chlorophenyl phenyl ether | 42 | 75-166 | 71-142 | 63-194 | |
| 240 | 4-chlorophenyl phenyl ether-d5 | 52 | 40-161 | 19-325 | 57-175 | 29-212 |
| 376 | Chrysene | 51 | 59-186 | 70-142 | 48-221 | |
| 276 | Chrysene-d12 | 69 | 33-219 | 13-512 | 24-411 | 23-290 |
| 713 | p-cymene (Appendix C) | 18 | 76-140 | 79-127 | 72-147 | |
| 613 | p-cymene-d14 | 67 | ns-359 | ns-ns | 66-152 | ns-468 |
| 082 | Dibenzo(a,h)anthracene* | 55 | 23-299 | 13-761 | 19-340 | |
| 705 | Dibenzofuran (Appendix C) | 20 | 85-136 | 73-136 | 79-146 | |
| 605 | Dibenzofuran-d8 | 31 | 47-136 | 28-220 | 66-150 | 39-160 |
| 704 | Dibenzothiophene (Synfuel) | 31 | 79-150 | 72-140 | 70-168 | |
| 604 | Dibenzothiophene-d8 | 31 | 48-130 | 29-215 | 69-145 | 40-156 |
| 368 | Di-n-butyl phthalate | 15 | 76-165 | 71-142 | 74-169 | |
| 268 | Di-n-butyl phthalate-d4 | 23 | 23-195 | 13-346 | 52-192 | 22-209 |
| 325 | 1,2-dichlorobenzene | 17 | 73-146 | 74-135 | 70-152 | |
| 225 | 1,2-dichlorobenzene-d4 | 35 | 14-212 | ns-494 | 61-164 | 11-247 |
| 326 | 1,3-dichlorobenzene | 43 | 63-201 | 65-154 | 55-225 | |
| 226 | 1,3-dichlorobenzene-d4 | 48 | 13-203 | ns-550 | 52-192 | ns-260 |
| 327 | 1,4-dichlorobenzene | 42 | 61-194 | 62-161 | 53-219 | |
| 227 | 1,4-dichlorobenzene-d4 | 48 | 15-193 | ns-474 | 65-153 | 11-245 |
| 328 | 3,3′-dichlorobenzidine | 26 | 68-174 | 77-130 | 64-185 | |
| 228 | 3,3′-dichlorobenzidine-d6 | 80 | ns-562 | ns-ns | 18-558 | ns-ns |
| 331 | 2,4-dichlorophenol | 12 | 85-131 | 67-149 | 83-135 | |
| 231 | 2,4-dichlorophenol-d3 | 28 | 38-164 | 24-260 | 64-157 | 34-182 |
| 370 | Diethyl phthalate | 44 | 75-196 | 74-135 | 65-222 | |
| 270 | Diethyl phthalate-d4 | 78 | ns-260 | ns-ns | 47-211 | ns-ns |
| 334 | 2,4-dimethylphenol | 13 | 62-153 | 67-150 | 60-156 | |
| 234 | 2,4-dimethylphenol-d3 | 22 | 15-228 | ns-449 | 58-172 | 14-242 |
| 371 | Dimethyl phthalate | 36 | 74-188 | 73-137 | 67-207 | |
| 271 | Dimethyl phthalate-d4 | 108 | ns-640 | ns-ns | 50-201 | ns-ns |
| 359 | 2,4-dinitrophenol | 18 | 72-134 | 75-133 | 68-141 | |
| 259 | 2,4-dinitrophenol-d3 | 66 | 22-308 | ns-ns | 39-256 | 17-378 |
| 335 | 2,4-dinitrotoluene | 18 | 75-158 | 79-127 | 72-164 | |
| 235 | 2,4-dinitrotoluene-d3 | 37 | 22-245 | 10-514 | 53-187 | 19-275 |
| 336 | 2,6-dinitrotoluene | 30 | 80-141 | 55-183 | 70-159 | |
| 236 | 2,6-dinitrotoluene-d3 | 59 | 44-184 | 17-442 | 36-278 | 31-250 |
| 369 | Di-n-octyl phthalate | 16 | 77-161 | 71-140 | 74-166 | |
| 269 | Di-n-octyl phthalate-d4 | 46 | 12-383 | ns-ns | 21-467 | 10-433 |
| 707 | Diphenylamine (Appendix C) | 45 | 58-205 | 57-176 | 51-231 | |
| 607 | Diphenylamine-d10 | 42 | 27-206 | 11-488 | 59-169 | 21-249 |
| 708 | Diphenyl ether (Appendix C) | 19 | 82-136 | 83-120 | 77-144 | |
| 608 | Diphenyl ether-d10 | 37 | 36-155 | 19-281 | 77-129 | 29-186 |
| 337 | 1,2-diphenylhydrazine | 73 | 49-308 | 75-134 | 40-360 | |
| 237 | 1,2-diphenylhydrazine-d10 | 35 | 31-173 | 17-316 | 58-174 | 26-200 |
| 339 | Fluoranthene | 33 | 71-177 | 67-149 | 64-194 | |
| 239 | Fluoranthene-d10 | 35 | 36-161 | 20-278 | 47-215 | 30-187 |
| 380 | Fluorene | 29 | 81-132 | 74-135 | 70-151 | |
| 280 | Fluorene-d10 | 43 | 51-131 | 27-238 | 61-164 | 38-172 |
| 309 | Hexachlorobenzene | 16 | 90-124 | 78-128 | 85-132 | |
| 209 | Hexachlorobenzene-13C6 | 81 | 36-228 | 13-595 | 38-265 | 23-321 |
| 352 | hexachlorobutadiene | 56 | 51-251 | 74-135 | 43-287 | |
| 252 | hexachlorobutadiene-13C4 | 63 | ns-316 | ns-ns | 68-148 | ns-413 |
| 312 | hexachloroethane | 227 | 21-ns | 71-141 | 13-ns | |
| 212 | hexachloroethane-13C1 | 77 | ns-400 | ns-ns | 47-212 | ns-563 |
| 353 | hexachlorocyclopentadiene | 15 | 69-144 | 77-129 | 67-148 | |
| 253 | hexachlorocyclopentadiene-13C4 | 60 | ns-ns | ns-ns | 47-211 | ns-ns |
| 083 | ideno(1,2,3-cd)pyrene* | 55 | 23-299 | 13-761 | 19-340 | |
| 354 | isophorone | 25 | 76-156 | 70-142 | 70-168 | |
| 254 | isophorone-d8 | 23 | 49-133 | 33-193 | 52-194 | 44-147 |
| 360 | 2-methyl-4,6-dinitrophenol | 19 | 77-133 | 69-145 | 72-142 | |
| 260 | 2-methyl-4,6-dinitrophenol-d2 | 64 | 36-247 | 16-527 | 56-177 | 28-307 |
| 355 | naphthalene | 20 | 80-139 | 73-137 | 75-149 | |
| 255 | naphthalene-d8 | 39 | 28-157 | 14-305 | 71-141 | 22-192 |
| 702 | B-naphthylamine (Appendix C) | 49 | 10-ns | 39-256 | ns-ns | |
| 602 | B-naphthylamine-d7 | 33 | ns-ns | ns-ns | 44-230 | ns-ns |
| 356 | nitrobenzene | 25 | 69-161 | 85-115 | 65-169 | |
| 256 | nitrobenzene-d5 | 28 | 18-265 | ns-ns | 46-219 | 15-314 |
| 357 | 2-nitrophenol | 15 | 78-140 | 77-129 | 75-145 | |
| 257 | 2-nitrophenol-d4 | 23 | 41-145 | 27-217 | 61-163 | 37-158 |
| 358 | 4-nitrophenol | 42 | 62-146 | 55-183 | 51-175 | |
| 258 | 4-nitrophenol-d4 | 188 | 14-398 | ns-ns | 35-287 | ns-ns |
| 061 | N-nitrosodimethylamile* | 198 | 21-472 | 40-249 | 12-807 | |
| 063 | N-nitrosodi-n-proplyamine* | 198 | 21-472 | 40-249 | 12-807 | |
| 362 | N-nitrosodiphenylamine | 45 | 65-142 | 68-148 | 53-173 | |
| 262 | N-nitrosodiphenylamine-d6 | 37 | 54-126 | 26-256 | 59-170 | 40-166 |
| 364 | pentachlorophenol | 21 | 76-140 | 77-130 | 71-150 | |
| 264 | pentachlorophenol-13C6 | 49 | 37-212 | 18-412 | 42-237 | 29-254 |
| 381 | phenanthrene | 13 | 93-119 | 75-133 | 87-126 | |
| 281 | phenanthrene-d10 | 40 | 45-130 | 24-241 | 67-149 | 34-168 |
| 365 | phenol | 36 | 77-127 | 65-155 | 62-154 | |
| 265 | phenol-d5 | 161 | 21-210 | ns-ns | 48-208 | ns-ns |
| 703 | a-picoline (Synfuel) | 38 | 59-149 | 60-165 | 50-174 | |
| 603 | a-picoline-d7 | 138 | 11-380 | ns-ns | 31-324 | ns-608 |
| 384 | pyrene | 19 | 76-152 | 76-132 | 72-159 | |
| 284 | pyrene-d10 | 29 | 32-176 | 18-303 | 48-210 | 28-196 |
| 710 | styrene (Appendix C) | 42 | 53-221 | 65-153 | 48-244 | |
| 610 | styrene-d5 | 49 | ns-281 | ns-ns | 44-228 | ns-348 |
| 709 | a-terpineol (Appendix C) | 44 | 42-234 | 54-186 | 38-258 | |
| 609 | a-terpineol-d3 | 48 | 22-292 | ns-672 | 20-502 | 18-339 |
| 529 | 1,2,3-trichlorobenzene (4c)* | 69 | 15-229 | 60-167 | 11-297 | |
| 308 | 1,2,4-trichlorobenzene | 19 | 82-136 | 78-128 | 77-144 | |
| 208 | 1,2,4-trichlorobenzene-d3 | 57 | 15-212 | ns-592 | 61-163 | 10-282 |
| 530 | 2,3,6-trichlorophenol (4c)* | 30 | 58-137 | 56-180 | 51-153 | |
| 531 | 2,4,5-trichlorophenol (4c)* | 30 | 58-137 | 56-180 | 51-153 | |
| 321 | 2,4,6-trichlorophenol | 57 | 59-205 | 81-123 | 48-244 | |
| 221 | 2,4,6-trichlorophenol-d2 | 47 | 43-183 | 21-363 | 69-144 | 34-226 |
1 Reference numbers beginning with 0, 1 or 5 indicate a pollutant quantified by the internal standard method; reference numbers beginning with 2 or 6 indicate a labeled compound quantified by the internal standard method; reference numbers beginning with 3 or 7 indicate a pollutant quantified by isotope dilution.
* Measured by internal standard; specification derived from related compound.
ns = no specification; limit is outside the range that can be measured reliably.
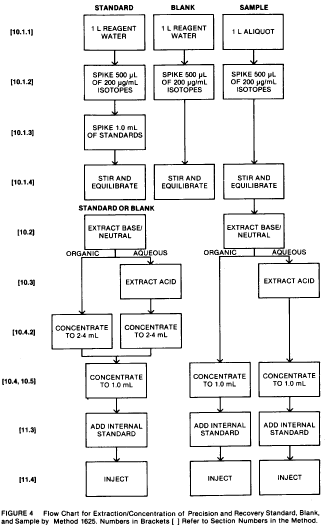
 Attachment 1 to
Method 1625 Introduction
Attachment 1 to
Method 1625 Introduction
To support measurement of several semivolatile pollutants, EPA has developed this attachment to EPA Method 1625B. 1 The modifications listed in this attachment are approved only for monitoring wastestreams from the Centralized Waste Treatment Point Source Category (40 CFR part 437) and the Landfills Point Source Category (40 CFR part 445). EPA Method 1625B (the Method) employs sample extraction with methylene chloride followed by analysis of the extract using capillary column gas chromatography-mass spectrometry (GC/MS). This attachment addresses the addition of the semivolatile pollutants listed in Tables 1 and 2 to all applicable standard, stock, and spiking solutions utilized for the determination of semivolatile organic compounds by EPA Method 1625B.
1 EPA Method 1625 Revision B, Semivolatile Organic Compounds by Isotope Dilution GC/MS, 40 CFR part 136, appendix A.
1.0 EPA METHOD 1625 REVISION B MODIFICATION SUMMARYThe additional semivolatile organic compounds listed in Tables 1 and 2 are added to all applicable calibration, spiking, and other solutions utilized in the determination of semivolatile compounds by EPA Method 1625. The instrument is to be calibrated with these compounds, and all procedures and quality control tests described in the Method must be performed.
2.0 SECTION MODIFICATIONS Note:All section and figure numbers in this Attachment reference section and figure numbers in EPA Method 1625 Revision B unless noted otherwise. Sections not listed here remain unchanged.
Section 6.7 The stock standard solutions described in this section are modified such that the analytes in Tables 1 and 2 of this attachment are required in addition to those specified in the Method. Section 6.8 The labeled compound spiking solution in this section is modified to include the labeled compounds listed in Tables 5 and 6 of this attachment. Section 6.9 The secondary standard is modified to include the additional analytes listed in Tables 1 and 2 of this attachment. Section 6.12 The solutions for obtaining authentic mass spectra are to include all additional analytes listed in Tables 1 and 2 of this attachment. Section 6.13 The calibration solutions are modified to include the analytes listed in Tables 1 and 2 and the labeled compounds listed in Tables 5 and 6 of this attachment. Section 6.14 The precision and recovery standard is modified to include the analytes listed in Tables 1 and 2 and the labeled compounds listed in Tables 5 and 6 of this attachment. Section 6.15 The solutions containing the additional analytes listed in Tables 1 and 2 of this attachment are to be analyzed for stability. Section 7.2.1 This section is modified to include the analytes listed in Tables 1 and 2 and the labeled compounds listed in Tables 5 and 6 of this attachment. Section 7.4.5 This section is modified to include the analytes listed in Tables 1 and 2 and the labeled compounds listed in Tables 5 and 6 in the calibration. Section 8.2 The initial precision and recovery (IPR) requirements are modified to include the analytes listed in Tables 1 and 2 and the labeled compounds listed in Tables 5 and 6 of this attachment. Additional IPR performance criteria are supplied in Table 7 of this attachment. Section 8.3 The labeled compounds listed in Tables 3 and 4 of this attachment are to be included in the method performance tests. Additional method performance criteria are supplied in Table 7 of this attachment. Section 8.5.2 The acceptance criteria for blanks includes the analytes listed in Tables 1 and 2 of this attachment. Section 10.1.2 The labeled compound solution must include the labeled compounds listed in Tables 5 and 6 of this attachment. Section 10.1.3 The precision and recovery standard must include the analytes listed in Tables 1 and 2 and the labeled compounds listed in Tables 5 and 6 of this attachment. Section 12.5 Additional QC requirements for calibration verification are supplied in Table 7 of this attachment. Section 12.7 Additional QC requirements for ongoing precision and recovery are supplied in Table 7 of this attachment.Table 1 - Base/Neutral Extractable Compounds
| Compound | Pollutant | |
|---|---|---|
| CAS Registry |
EPA-EGD | |
| acetophenone 1 | 98-86-2 | 758 |
| aniline 2 | 62-53-3 | 757 |
| -2,3-dichloroaniline 1 | 608-27-5 | 578 |
| -o-cresol 1 | 95-48-7 | 771 |
| pyridine 2 | 110-86-1 | 1330 |
CAS = Chemical Abstracts Registry.
EGD = Effluent Guidelines Division.
1 Analysis of this pollutant is approved only for the Centralized Waste Treatment industry.
2 Analysis of this pollutant is approved only for the Centralized Waste Treatment and Landfills industries.
Table 2 - Acid Extractable Compounds
| Compound | Pollutant | |
|---|---|---|
| CAS Registry |
EPA-EGD | |
| p-cresol 1 | 106-44-5 | 1744 |
CAS = Chemical Abstracts Registry.
EGD = Effluent Guidelines Division.
1 Analysis of this pollutant is approved only for the Centralized Waste Treatment and Landfills industries.
Table 3 - Gas Chromatography 1 of Base/Neutral Extractable Compounds
| EGD No. | Compound | Retention time 2 | Minimum level 3 (µg/L) |
EGD No. | Compound | Retention time 2 | Minimum level
3 (µg/L) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean (sec) |
EGD Ref | Relative | Mean (sec) |
EGD Ref | Relative | ||||||
| 758 | acetophenone 4 | 818 | 658 | 1.003-1.005 | 10 | ||||||
| 757 | aniline 5 | 694 | 657 | 0.994-1.023 | 10 | ||||||
| 578 | 2,3-dichloroaniline 4 | 1160 | 164 | 1.003-1.007 | 10 | ||||||
| 771 | o-cresol 4 | 814 | 671 | 1.005-1.009 | 10 | ||||||
| 1330 | pyridine 5 | 378 | 1230 | 1.005-1.011 | 10 | ||||||
EGD = Effluent Guidelines Division.
1 The data presented in this table were obtained under the chromatographic conditions given in the footnote to Table 3 of EPA Method 1625B.
2 Retention times are approximate and are intended to be consistent with the retention times for the analytes in EPA Method 1625B.
3 See the definition in footnote 2 to Table 3 of EPA Method 1625B.
4 Analysis of this pollutant is approved only for the Centralized Waste Treatment industry.
5 Analysis of this pollutant is approved only for the Centralized Waste Treatment and Landfills industries.
Table 4 - Gas Chromatography 1 of Acid Extractable Compounds
| EGD No. | Compound | Retention time 2 | Minimum level (µ/L) 3 |
||
|---|---|---|---|---|---|
| Mean (sec) |
EGD Ref | Relative | |||
| 1744 | p-cresol 4 | 834 | 1644 | 1.004-1.008 | 20 |
EGD = Effluent Guidelines Division.
1 The data presented in this table were obtained under the chromatographic conditions given in the footnote to Table 4 of EPA Method 1625B.
2 Retention times are approximate and are intended to be consistent with the retention times for the analytes in EPA Method 1625B.
3 See the definition in footnote 2 to Table 4 of EPA Method 1625B.
4 Analysis of this pollutant is approved only for the Centralized Waste Treatment and Landfills industries.
Table 5 - Base/Neutral Extractable Compound Characteristic m/z's
| Compound | Labeled Analog | Primary m/z 1 |
|---|---|---|
| acetophenone 2 | d5 | 105/110 |
| aniline 3 | d7 | 93/100 |
| o-cresol 2 | d7 | 108/116 |
| 2,3-dichloroaniline 2 | n/a | 161 |
| pyridine 3 | d5 | 79/84 |
m/z = mass to charge ratio.
1 Native/labeled.
2 Analysis of this pollutant is approved only for the Centralized Waste Treatment industry.
3 Analysis of this pollutant is approved only for the Centralized Waste Treatment and Landfills industries.
Table 6 - Acid Extractable Compound Characteristic m/z's
| Compound | Labeled Analog | Primary m/z 1 |
|---|---|---|
| p-cresol 2 | d7 | 108/116 |
m/z = mass to charge ratio.
1 Native/labeled.
2 Analysis of this pollutant is approved only for the Centralized Waste Treatment and Landfills industries.
Table 7 - Acceptance Criteria for Performance Tests
| EGD No. | Compound | Acceptance criteria | Calibration
verification sec. 12.5 µg/mL) |
On-going accuracy
sec. 12.7 R (µg/L) |
||
|---|---|---|---|---|---|---|
| Initial precision
and accuracy section 8.2 (µg/L) |
Labeled compound
recovery sec. 8.3 and 14.2 P (percent) |
|||||
| s (µg/L) |
X | |||||
| 758 | acetophenone 1 | 34 | 44-167 | 85-115 | 45-162 | |
| 658 | acetophenone-d 5 1 | 51 | 23-254 | 45-162 | 85-115 | 22-264 |
| 757 | aniline 2 | 32 | 30-171 | 85-115 | 33-154 | |
| 657 | aniline-d 7 2 | 71 | 15-278 | 33-154 | 85-115 | 12-344 |
| 771 | o-cresol 1 | 40 | 31-226 | 85-115 | 35-196 | |
| 671 | o-cresol-d 7 1 | 23 | 30-146 | 35-196 | 85-115 | 31-142 |
| 1744 | p-cresol 2 | 59 | 54-140 | 85-115 | 37-203 | |
| 1644 | p-cresol-d7 2 | 22 | 11-618 | 37-203 | 85-115 | 16-415 |
| 578 | 2,3-dichloroaniline 1 | 13 | 40-160 | 85-115 | 44-144 | |
| 1330 | pyridine 2 | 28 | 10-421 | 83-117 | 18-238 | |
| 1230 | pyridine-d 5 2 | ns | 7-392 | 19-238 | 85-115 | 4-621 |
s = Standard deviation of four recovery measurements.
X = Average recovery for four recovery measurements.
EGD = Effluent Guidelines Division.
ns = no specification; limit is outside the range that can be measured reliably.
1 Analysis of this pollutant is approved only for the Centralized Waste Treatment industry.
2 Analysis of this pollutant is approved only for the Centralized Waste Treatment and Landfills industries.