Title 40
SECTION 799.9365
799.9365 TSCA combined repeated dose toxicity study with the reproduction/developmental toxicity screening test.
§ 799.9365 TSCA combined repeated dose toxicity study with the reproduction/developmental toxicity screening test.(a) Scope - (1) Applicability. This section is intended to meet testing requirements of the Toxic Substances Control Act (TSCA) (15 U.S.C. 2601).
(2) Source. The source material used in developing this TSCA test guideline is the Office of Prevention, Pesticides and Toxic Substances (OPPTS) harmonized test guideline 870.3650 (July 2000, final guidelines). This source is available at the address in paragraph (h) of this section.
(b) Purpose. (1) This screening test provides limited information on systemic toxicity, neurotoxicity, and/or immunotoxicity following repeated exposure over a limited time period. In addition, it can be used to provide initial information on possible effects on male and female reproductive performance such as gonadal function, mating behavior, conception, development of the conceptus, and parturition. It is not an alternative to, nor does it replace, the existing test guidelines in §§ 799.9370, 799.9380, 799.9620, and 799.9780 of this part.
(2) This test does not provide complete information on all aspects of reproduction and development. In particular, it offers only limited means of detecting postnatal manifestations of prenatal exposure, or effects that may be induced during postnatal exposure. Due (amongst other reasons) to the selectivity of the end points, and the short duration of the study, this method will not provide evidence for definite claims of no reproduction/developmental effects.
(3) This test can be used to provide initial information either at an early stage of assessing the toxicological properties of chemicals, or chemicals of high concern. It can also be used as part of a set of initial screening tests for existing chemicals for which little or no toxicological information is available or when otherwise considered relevant. It also can serve as an alternative to conducting two separate screening tests for repeated dose toxicity as described in § 799.9305 of this part and reproductive/developmental toxicity as described in § 799.9355 of this part.
(c) Definitions. The definitions in section 3 of TSCA and in 40 CFR Part 792 - Good Laboratory Practice Standards apply to this section. The following definitions also apply to this section.
Dosage is a general term comprising dose, its frequency and the duration of dosing.
Dose is the amount of test substance administered. Dose is expressed as weight (g, gm) or as weight of test substance per unit weight of test animal (e.g., mg/kg), or as constant dietary concentration (parts per million (ppm)).
No-observed-effects level (NOEL) is the maximum dose used in a study which produces no adverse effects. The NOEL is expressed in terms of the weight of a test substance given daily per unit weight of test animal (milligrams per kilogram per day).
(d) Principle of the test. (1) The test substance must be administered in graduated doses to several groups of males and females. Males should be dosed for a minimum of 4 weeks, up to and including the day before scheduled sacrifice (this includes a minimum of 2 weeks prior to mating, during the mating period and, approximately, 2 weeks post mating). In view of the limited pre-mating dosing period in males, fertility may not be a particularly sensitive indicator of testicular toxicity. Therefore, a detailed histological examination of the testes is essential. The combination of a pre-mating dosing period of 2 weeks and subsequent mating/fertility observations with an overall dosing period of at least 4 weeks, followed by detailed histopathology of the male gonads, is considered sufficient to enable detection of the majority of effects on male fertility and spermatogenesis.
(2) Females should be dosed throughout the study. This includes 2 weeks prior to mating (with the objective of covering at least two complete oestrous cycles), the variable time to conception, the duration of pregnancy and at least 4 days after delivery, up to and including the day before scheduled sacrifice.
(3) Duration of study, following acclimatization, is dependent on the female performance and is approximately 54 days, (at least 14 days pre-mating, (up to) 14 days mating, 22 days gestation, 4 days lactation).
(4) During the period of administration, the animals are observed closely each day for signs of toxicity. Animals which die or are sacrificed during the test are necropsied and, at the conclusion of the test, surviving animals are sacrificed and necropsied.
(e) Description of the method - (1) Selection of animal species. This test guideline is designed for use with the rat. If other species are used, appropriate modifications will be necessary. Strains with low fecundity or well-known high incidence of developmental defects should not be used. Healthy virgin animals, not subjected to previous experimental procedures, should be used. The test animals should be characterised as to species, strain, sex, weight and/or age. At the commencement of the study the weight variation of animals used should be minimal and not exceed ±20% of the mean weight of each sex. Where the study is conducted as a preliminary study to a long-term or a full-generation study, preferably animals from the same strain and source should be used in both studies.
(2) Housing and feeding conditions. (i) The temperature in the experimental animal room should be 22 °C (±3°). The relative humidity should be at least 30% and preferably not exceed 70% other than during room cleaning. Lighting should be artificial, the sequence being 12 hours light, 12 hours dark. For feeding, conventional laboratory diets may be used with an unlimited supply of drinking water. The choice of diet may be influenced by the need to ensure a suitable admixture of a test substance when administered by this method.
(ii) Animals may be housed individually or be caged in small groups of the same sex; for group caging, no more than five animals should be housed per cage. Mating procedures should be carried out in cages suitable for the purpose. Pregnant females should be caged individually and provided with nesting materials.
(3) Preparation of the animals. Healthy young adult animals must be randomised and assigned to the treatment groups and cages. Cages should be arranged in such a way that possible effects due to cage placements are minimized. The animals must be uniquely identified and kept in their cages for at least 5 days prior to the start of the study to allow for acclimatisation to the laboratory conditions.
(4) Preparation of doses. (i) It is recommended that the test substance be administered orally unless other routes of administration are considered more appropriate. When the oral route is selected, the test compound is usually administered by gavage; however, alternatively, test compounds may also be administered via the diet or drinking water.
(ii) Where necessary, the test substance is dissolved or suspended in a suitable vehicle. It is recommended that, wherever possible, the use of an aqueous solution/suspension be considered first, followed by consideration of a solution/emulsion in oil (e.g., corn oil) and then by possible solution in other vehicles. For non-aqueous vehicles the toxic characteristics of the vehicle must be known. The stability of the test substance in the vehicle should be determined.
(f) Procedure - (1) Number and sex of animals. It is recommended that each group be started with at least 10 animals of each sex. Except in the case of marked toxic effects, it is expected that this will provide at least eight pregnant females per group which normally is the minimum acceptable number of pregnant females per group. The objective is to produce enough pregnancies and offspring to assure a meaningful evaluation of the potential of the substance to affect fertility, pregnancy, maternal and suckling behaviour, and growth and development of the F1 offspring from conception to day 4 post-partum. If interim sacrifices are planned, the number should be increased by the number of animals scheduled to be sacrificed before the completion of the study. Consideration should be given to an additional satellite group of five animals per sex in the control and the top dose group for observation of reversibility, persistence or delayed occurrence of systemic toxic effects, for at least 14 days post treatment. Animals of the satellite groups must not be mated and, consequently, must not used for the assessment of reproduction/developmental toxicity.
(2) Dosage. (i) Generally, at least three test groups and a control group should be used. If there are no suitable general toxicity data available, a range finding study may be performed to aid the determination of the doses to be used. Except for treatment with the test substance, animals in the control group should be handled in an identical manner to the test group subjects. If a vehicle is used in administering the test substance, the control group should receive the vehicle in the highest volume used.
(ii) Dose levels should be selected taking into account any existing toxicity and (toxico-) kinetic data available for the test compound or related materials. It should also be taken into account that there may be differences in sensitivity between pregnant and non-pregnant animals. The highest dose level should be chosen with the aim of inducing toxic effects but not death nor obvious suffering. Thereafter, a descending sequence of dose levels should be selected with a view to demonstrating any dosage related response and no adverse effects at the lowest dose level. Two- to four-fold intervals are frequently optimum and addition of a fourth test group is often preferable to using very large intervals (e.g., more than a factor of 10) between dosages.
(3) Limit test. If an oral study at 1-dose level of at least 1000 mg/kg body weight/day or, for dietary administration, an equivalent percentage in the diet, or drinking water (based upon body weight determinations), using the procedures described for this study, produces no observable toxic effects and if toxicity would not be expected based upon data from structurally related compounds, then a full study using several dose levels may not be considered necessary. The limit test applies except when human exposure indicates the need for a higher dose level to be used. For other types of administration, such as inhalation or dermal application, the physical chemical properties of the test substance often may dictate the maximum attainable exposure.
(4) Administration of doses. (i) The animals are dosed with the test substance daily for 7 days a week. When the test substance is administered by gavage, this should be done in a single dose to the animals using a stomach tube or a suitable intubation cannula. The maximum volume of liquid that can be administered at one time depends on the size of the test animal. The volume should not exceed 1 ml/100 g body weight, except in the case of aqueous solutions where 2 ml/100 g body weight may be used. Except for irritating or corrosive substances which will normally reveal exacerbated effects with higher concentrations, variability in test volume should be minimized by adjusting the concentration to ensure a constant volume at all dose levels.
(ii) For substances administered via the diet or drinking water, it is important to ensure that the quantities of the test substance involved do not interfere with normal nutrition or water balance. When the test substance is administered in the diet either a constant dietary concentration (parts per million (ppm)) or a constant dose level in terms of the animals' body weight may be used; the alternative used must be specified. For a substance administered by gavage, the dose should be given at similar times each day, and adjusted at least weekly to maintain a constant dose level in terms of animal body weight.
(5) Experimental schedule. (i) Dosing of both sexes should begin 2 weeks prior to mating, after they have been acclimatized for at least 5 days. The study should be scheduled in such a way that mating begins soon after the animals have attained full sexual maturity. This may vary slightly for different strains of rats in different laboratories, e.g., Sprague Dawley rats 10 weeks of age, Wistar rats about 12 weeks of age. Dams with offspring should be sacrificed on day 4 post-partum, or shortly thereafter. In order to allow for overnight fasting of dams prior to blood collection (if this option is preferred), dams and their offspring need not necessarily be sacrificed on the same day. The day of birth (viz. when parturition is complete) is defined as day 0 post-partum. Females showing no-evidence of copulation are sacrificed 24-26 days after the last day of the mating period. Dosing is continued in both sexes during the mating period. Males should further be dosed after the mating period at least until the minimum total dosing period of 28 days has been completed. They are then sacrificed, or, alternatively, are retained and continued to be dosed for the possible conduction of a second mating if considered appropriate.
(ii) Daily dosing of the parental females should continue throughout pregnancy and at least up to, and including, day 3 post-partum or the day before sacrifice. For studies where the test substance is administered by inhalation or by the dermal route, dosing should be continued at least up to, and including, day 19 of gestation.
(iii) Animals in a satellite group scheduled for follow-up observations, if included, must not mated. They should be kept at least for a further 14 days after the first scheduled sacrifice of dams, without treatment to detect delayed occurrence, or persistence of, or recovery from toxic effects.
(iv) The experimental schedule is given in the following figure 1.
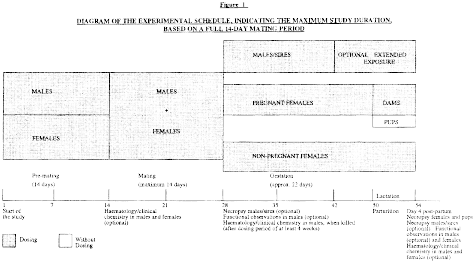
(6) Mating procedure. Normally, 1:1 (one male to one female) matings should be used in this study. Exceptions can arise in the case of occasional deaths of males. The female should be placed with the same male until pregnancy occurs or 2 weeks have elapsed. Each morning the females should be examined for the presence of sperm or a vaginal plug. Day 0 of pregnancy is defined as the day a vaginal plug or sperm is found. In case pairing was unsuccessful, re-mating of females with proven males of the same group could be considered.
(7) Observations. (i) General clinical observations should be made at least once a day, preferably at the same time(s) each day and considering the peak period of anticipated effects after dosing. The health condition of the animals should be recorded. At least twice daily all animals must be observed for morbidity and mortality.
(ii) Once before the first exposure (to allow for within-subject comparisons), and at least once a week thereafter, detailed clinical observations should be made in all animals. These observations should be made outside the home cage in a standard arena and preferably at the same time, each day. They should be carefully recorded; preferably using scoring systems, explicitly defined by the testing laboratory. Effort should be made to ensure that variations in the test conditions are minimal and that observations are preferably conducted by observers unaware of the treatment. Signs noted should include, but not be limited to, changes in skin, fur, eyes, mucous membranes, occurrence of secretions and excretions and autonomic activity (e.g., lacrimation, piloerection, pupil size, unusual respiratory pattern). Changes in gait, posture and response to handling as well as the presence of clonic or tonic movements, stereotypies (e.g., excessive grooming, repetitive circling), difficult or prolonged parturition or bizarre behaviour (e.g., self-mutilation, walking backwards) should also be recorded.
(iii) At one time during the study, sensory reactivity to stimuli of different modalities (e.g., auditory, visual and proprioceptive stimuli) assessment of grip strength and motor activity assessment should be conducted in five males and five females, randomly selected from each group. Further details of the procedures that could be followed are given in the respective references. However, alternative procedures than those referenced could also be used. In males, these functional observations should be made towards the end of their dosing period, shortly before scheduled sacrifice but before blood sampling for hematology or clinical chemistry. Females should be in a physiologically similar state during these functional tests and should preferably be tested during lactation, shortly before scheduled sacrifice. In order to avoid hypothermia of pups, dams should be removed from the pups for not more than 30 to 40 minutes. Examples of procedures for observation are described in the references in paragraphs (h)(3), (h)(4), (h)(5), (h)(6), and (h)(7) of this section.
(iv) Functional observations made once towards the end of the study may be omitted when the study is conducted as a preliminary study to a subsequent subchronic (90-day) or long-term study. In that case, the functional observations should be included in this follow-up study. On the other hand, the availability of data on functional observations from this repeated dose study may enhance the ability to select dose levels for a subsequent subchronic or long-term study.
(v) Functional observations may also be omitted for groups that otherwise reveal signs of toxicity to an extent that would significantly interfere with the functional test performance.
(vi) The duration of gestation should be recorded and is calculated from day 0 of pregnancy. Each litter should be examined as soon as possible after delivery to establish the number and sex of pups, stillbirths, live births, runts (pups that are significantly smaller than corresponding control pups), and the presence of gross abnormalities.
(vii) Live pups should be counted and sexed and litters weighed within 24 hours of parturition (day 0 or 1 post-partum) and on day 4 post-partum. In addition to the observations on parental animals, described by paragraphs (f)(7)(ii) and (f)(7)(iii) of this section, any abnormal behaviour of the offspring should be recorded.
(8) Body weight and food/water consumption. (i) Males and females should be weighed on the first day of dosing, at least weekly thereafter, and at termination. During pregnancy, females should be weighed on days 0, 7, 14 and 20 and within 24 hours of parturition (day 0 or 1 post-partum), and day 4 post-partum. These observations should be reported individually for each adult animal.
(ii) During pre-mating, pregnancy and lactation, food consumption should be measured at least weekly. The measurement of food consumption during mating is optional. Water consumption during these periods should also be measured, when the test substance is administered by that medium.
(9) Hematology. (i) Once during the study, the following hematological examinations should be made in five males and five females randomly selected from each group: hematocrit, hemoglobin concentration, erythrocyte count, total and differential leucocyte count, platelet count and a measure of blood clotting time/potential.
(ii) Blood samples should be taken from a named site. Females should be in a physiologically similar state during sampling. In order to avoid practical difficulties related to the variability in the onset of gestation, blood collection in females may be done at the end of the pre-mating period as an alternative to sampling just prior to, or as part of, the procedure for sacrificing the animals. Blood samples of males should preferably be taken just prior to, or as part of, the procedure for sacrificing the animals. Alternatively, blood collection in males may also be done at the end of the pre-mating period when this time point was preferred for females.
(iii) Blood samples should be stored under appropriate conditions.
(10) Clinical biochemistry. (i) Clinical biochemistry determinations to investigate major toxic effects in tissues and, specifically, effects on kidney and liver, should be performed on blood samples obtained from the selected five males and five females of each group. Overnight fasting of the animals prior to blood sampling is recommended 1 . Investigations of plasma or serum must include sodium, potassium, glucose, total cholesterol, urea, creatinine, total protein and albumin, at least two enzymes indicative of hepatocellular effects (such as alanine aminotransferase, aspartate aminotransferase and sorbitol dehydrogenase) and bile acids. Measurements of additional enzymes (of hepatic or other origin) may provide useful information under certain circumstances.
1 For a number of measurements in serum and plasma, most notably for glucose, overnight fasting would be preferable. The major reason for this preference is that the increased variability which would inevitably result from non-fasting, would tend to mask more subtle effects and make interpretation difficult. On the other hand, however, overnight fasting may interfere with the general metabolism of the (pregnant) animals, disturbs lactation and nursing behaviour, and, particularly in feeding studies, may disturb the daily exposure to the test substance. If overnight fasting is adopted, clinical biochemical determinations should be performed after the conduct of functional observations in week 4 of the study.
(ii) Optionally, the following urinalysis determinations could be performed in five randomly selected males of each group during the last week of the study using timed urine volume collection; appearance, volume, osmolality or specific gravity, pH, protein, glucose and blood or blood cells.
(iii) In addition, studies to investigate serum markers of general tissue damage should be considered. Other determinations that should be carried out if the known properties of the test substance may, or are suspected to, affect related metabolic profiles include calcium, phosphate, fasting triglycerides and fasting glucose, specific hormones, methemoglobin and cholinesterase. These need to be identified on a case-by-case basis.
(iv) Overall, there is a need for a flexible approach, depending on the observed and/or expected effect with a given compound.
(v) If historical baseline data are inadequate, consideration should be given to determination of hematological and clinical biochemistry variables before dosing commences.
(11) Pathology - (i) Gross necropsy. (A) All adult animals in the study must be subjected to a full, detailed gross necropsy which includes careful examination of the external surface of the body, all orifices, and the cranial, thoracic and abdominal cavities and their contents. Special attention should be paid to the organs of the reproductive system. The number of implantation sites should be recorded. Corpora lutea should be counted.
(B) The testes and epididymides of all adult males should be weighed and the ovaries, testes, epididymides, accessory sex organs, and all organs showing macroscopic lesions of all adult animals, should be preserved.
(C) In addition, for five adult males and females, randomly selected from each group, the liver, kidneys, adrenals, thymus, spleen, brain and heart should be trimmed of any adherent tissue, as appropriate and their wet weight taken as soon as possible after dissection to avoid drying. Of the selected males and females, the following tissues should also be preserved in the most appropriate fixation medium for both the type of tissue and the intended subsequent histopathological examination: all gross lesions, brain (representative regions including cerebrum, cerebellum and pons), spinal cord, stomach, small and large intestines (including Peyer's patches), liver, kidneys, adrenals, spleen, heart, thymus, thyroid, trachea and lungs (preserved by inflation with fixative and then immersion), uterus, urinary bladder, lymph nodes (preferably 1 lymph node covering the route of administration and another one distant from the route of administration to cover systemic effects), peripheral nerve (sciatic or tibial) preferably in close proximity to the muscle, and a section of bone marrow (or, alternatively, a fresh mounted marrow aspirate).
(D) Formalin fixation is not recommended for routine examination of testes and epididymides. An acceptable method is the use of Bouin's fixative for these tissues. The clinical and other findings may suggest the need to examine additional tissues. Also, any organs considered likely to be target organs based on the known properties of the test substance should be preserved.
(E) Dead pups and pups sacrificed at day 4 post-partum, or shortly thereafter, should, at least, be carefully examined externally for gross abnormalities.
(ii) Histopathology. (A) Full histopathology should be conducted on the preserved organs and tissues of the selected animals in the control and high dose groups and all gross lesions. These examinations should be extended to animals of other dosage groups if treatment-related changes are observed in the high dose group.
(B) Detailed testicular histopathological examination (e.g., using Bouin's fixative, paraffin embedding and transverse sections of 4-5 ±m thickness) should be conducted with special emphasis on stages of spermatogenesis and histopathology interstitial testicular cell structure. The evaluation should identify treatment-related effects such as retained spermatids, missing germ cell layers or types, multinucleated giant cells or sloughing of spermatogenic cells into the lumen (the specifications for the evaluation are discussed in paragraph (g)(2) of this section). Examination of the intact epididymis should include the caput, corpus, and cauda, which can be accomplished by evaluation of a longitudinal section. The epididymis should be evaluated for leukocyte infiltration, change in prevalence of cell types, aberrant cell types, and phagocytosis of sperm. Periodic acid-Schiff (PAS) and hematoxylin staining may be used for examination of the male reproductive organs. Histopathological examination of the ovary should detect qualitative depletion of the primordial follicle population.
(C) When a satellite group is used, histopathology should be performed on tissues and organs identified as showing effects in the treated groups.
(g) Data and reporting - (1) Data. Individual animal data should be provided. Additionally, all data should be summarised in tabular form, showing for each test group the number of animals at the start of the test, the number of animals found dead during the test or sacrificed for humane reasons, the time of any death or humane sacrifice, the number of fertile animals, the number of pregnant females, the number of animals showing signs of toxicity, a description of the signs of toxicity observed, including time of onset, duration, and severity of any toxic effects, the types of histopathological changes, and all relevant litter data.
(2) Evaluation of results. (i) The findings of this toxicity study should be evaluated in terms of the observed effects, necropsy and microscopic findings. The evaluation will include the relationship between the dose of the test substance and the presence or absence, incidence and severity of abnormalities, including gross lesions, identified target organs, infertility, clinical abnormalities, affected reproductive and litter performance, body weight changes, effects on mortality and any other toxic effects.
(ii) Because of the short period of treatment of the male, the histopathology of the testes and epididymides must be considered along with the fertility data, when assessing male reproduction effects. The use of historic control data on reproduction/development (e.g. for litter size) where available may also be useful as an aid to the interpretation of the study.
(iii) When possible, numerical results should be evaluated by an appropriate and general acceptable statistical method. The statistical methods should be selected during the design of the study. Due to the limited dimensions of the study, statistical analysis in the form of tests for “significance” are of limited value for many endpoints, especially reproductive endpoints. Some of the most widely used methods, especially parametric tests for measures of central tendency, are inappropriate. If statistical analyses are used then the method chosen should be appropriate for the distribution of the variable examined and be selected prior to the start of the study.
(3) Test report. The test report must include the following information:
(i) Test substance:
(A) Physical nature and, where relevant, physicochemical properties.
(B) Identification data.
(ii) Vehicle (if appropriate): Justification for choice of vehicle, if other than water.
(iii) Test animals:
(A) Species/strain used.
(B) Number, age and sex of animals.
(C) Source, housing conditions, diet, etc.
(D) Individual weights of animals at the start of the test.
(iv) Test conditions:
(A) Rationale for dose level selection.
(B) Details of test substance formulation/diet preparation, achieved concentration, stability and homogeneity of the preparation.
(C) Details of the administration of the test substance.
(D) Conversion from diet/drinking water test substance concentration (parts per mission (ppm)) to the actual dose (mg/kg body weight/day), if applicable.
(E) Details of food and water quality.
(v) Results (toxic response data by sex and dose):
(A) Time of death during the study or whether animals survived to termination.
(B) Nature, severity and duration of clinical observations (whether reversible or not).
(C) Body weight/body weight change data.
(D) Food consumption and water consumption, if applicable.
(E) Sensory activity, grip strength and motor activity assessments.
(F) Hematological tests with relevant baseline values,
(G) Clinical biochemistry tests with relevant baseline values.
(H) Effects on reproduction, including information on mating/precoital interval, fertility, fecundity and gestation duration.
(I) Effects on offspring, including number of pups born (live and dead), sex ratio, postnatal growth (pup weights) and survival (litter size), gross abnormalities and clinical observations during lactation.
(J) Body weight at termination and organ weight data for the parental animals.
(K) Necropsy data, including number of implantations and number of corpora lutea.
(L) Calculations of pre- and postimplantation loss.
(M) Detailed description of histopathological findings.
(N) Statistical treatment of results, where appropriate.
(vi) Discussion of results.
(vii) Conclusions.
(h) References. For additional background information on this test guideline, the following references should be consulted. These references are available at the addresses in § 700.17(b)(1) and (2) of this chapter.
(1) Mitsumori, K., Kodama, Y., Uchida, O., Takada, K., Saito, M. Naito, K., Tanaka, S., Kurokawa, Y., Usami, M., Kawashima, K., Yasuhara, K., Toyoda, K., Onodera, H., Furukawa, F., Takahashi, M. and Hayashi, Y., (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). Journal of Toxicology and Science, 19:141-149.
(2) Tanaka, S., Kawashima, K., Naito, K., Usami, M., Nakadate, M., Imaida, K., Takahashi, M., Hayashi, Y., Kurokawa, Y. and Tobe, M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundamental and Applied Toxicology, 18:89-95.
(3) Tupper D.E., Wallace R.B. (1980). Utility of the Neurologic Examination in Rats. Acta Neurobiological Exposure, 40:999-1003.
(4) Gad S.C. (1982). A Neuromuscular Screen for Use in Industrial Toxicology. Journal of Toxicology and Environmental Health, 9:691-704.
(5) Moser V.C., McDaniel K.M., Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicology and Applied Pharmacology, 108:267-283.
(6) Meyer O.A., Tilson H.A., Byrd W.C., Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehavorial Toxicology, 1:233-236.
(7) Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicology and Teratology 13:599-609.
[65 FR 78793, Dec. 15, 2000, as amended at 77 FR 42694, Aug. 3, 2012]