Title 40
SECTION 799.6786
799.6786 TSCA water solubility: Generator column method.
§ 799.6786 TSCA water solubility: Generator column method.(a) Scope - (1) Applicability. This section is intended to meet the testing requirements of the Toxic Substances Control Act (TSCA) (15 U.S.C. 2601).
(2) Source. The source material used in developing this TSCA test guideline is the Office of Pollution Prevention, Pesticides and Toxics (OPPTS) harmonized test guideline 830.7860 (March 1998, revised final guideline). The source is available at the address in paragraph (e) of this section.
(b) Introduction - (1) Purpose. (i) The water solubility of a chemical is defined as the equilibrium concentration of the chemical in a saturated aqueous solution at a given temperature and pressure. The aqueous phase solubility is an important factor in governing the movement, distribution, and rate of degradation of chemicals in the environment. Substances that are relatively water soluble are more likely to be widely distributed by the hydrologic cycle than those which are relatively insoluble. Furthermore, substances with higher water solubility are more likely to undergo microbial or chemical degradation in the environment because dissolution makes them “available” to interact and, therefore, react with other chemicals and microorganisms. Both the extent and rate of degradation via hydrolysis, photolysis, oxidation, reduction, and biodegradation depend on a chemical being soluble in water (i.e., homogeneous kinetics).
(ii) Water provides the medium in which many organisms live, and water is a major component of the internal environment of all living organisms (except for dormant stages of certain life forms). Even organisms which are adapted to life in a gaseous environment require water for normal functioning. Water is thus the medium through which most other chemicals are transported to and into living cells. As a result, the extent to which chemicals dissolve in water will be a major determinant for movement through the environment and entry into living systems.
(iii) The water solubility of a chemical also has an effect on its sorption into and desorption from soils and sediments, and on volatilization from aqueous media. The more soluble a chemical substance is, the less likely it is to sorb to soils and sediments and the less likely it is to volatilize from water. Finally, the design of most chemical tests and many ecological and health tests requires precise knowledge of the water solubility of the chemical to be tested.
(2) Definitions. The following definitions apply to this section.
Concentration (C) of a solution is the amount of solute in a given amount of solvent or solution and can be expressed as a weight/weight or weight/volume relationship. The conversion from a weight relationship to one of volume incorporates density as a factor. For dilute aqueous solutions, the density of the solvent is approximately equal to the density of the solution; thus, concentrations expressed in milligrams per liter (mg/L) are approximately equal to 10−3 g/10 3 g or parts per million (ppm); those expressed in micrograms per liter (µg/L) are approximately equal to 10−6 g/10 3 g or parts per billion (ppb). In addition, concentration can be expressed in terms of molarity, normality, molality, and mole fraction. For example, to convert from weight/volume to molarity molecular mass is incorporated as a factor.
Density is the mass of a unit volume of a material. It is a function of temperature, hence the temperature at which it is measured should be specified. For a solid, it is the density of the impermeable portion rather than the bulk density. For solids and liquids, suitable units of measurement are grams per cubic centimeter (g/cm 3). The density of a solution is the mass of a unit volume of the solution and suitable units of measurement are g/cm 3.
Extractor column is used to extract the solute from the saturated solutions produced by the generator column. After extraction onto a chromatographic support, the solute is eluted with a solvent/water mixture and subsequently analyzed by high-pressure liquid chromatography (HPLC), gas chromatography (GC), or any other suitable analytical procedure. A detailed description of the preparation of the extractor column is given in paragraph (c)(1)(i)(D) of this section.
Generator column is used to produce or generate saturated solutions of a solute in a solvent. The column, see figure 1 in paragraph (c)(1)(i)(A) of this section, is packed with a solid support coated with the solute, i.e., the organic compound whose solubility is to be determined. When water (the solvent) is pumped through the column, saturated solutions of the solute are generated. Preparation of the generator column is described in paragraph (c)(1)(i)(A) of this section.
Response factor (RF) is the solute concentration required to give a 1 unit area chromatographic peak or 1 unit output from the HPLC recording integrator at a particular recorder attenuation. The factor is required to convert from units of area to units of concentration. The determination of the RF is given in paragraph (c)(3)(ii)(B)(2) of this section.
Sample loop is a 1/16 inch (in) outer diameter (O.D.) (1.6 millimeter (mm)) stainless steel tube with an internal volume between 20 and 50 µL. The loop is attached to the sample injection valve of the HPLC and is used to inject standard solutions into the mobile phase of the HPLC when determining the RF for the recording integrator. The exact volume of the loop must be determined as described in paragraph (c)(3)(ii)(B)(1) of this section when the HPLC method is used.
Saturated solution is a solution in which the dissolved solute is in equilibrium with an excess of undissolved solute; or a solution in equilibrium such that at a fixed temperature and pressure, the concentration of the solute in the solution is at its maximum value and will not change even in the presence of an excess of solute.
Solution is a homogeneous mixture of two or more substances constituting a single phase.
(3) Principle of the test method. (i) This test method is based on the dynamic coupled column liquid chromatographic (DCCLC) technique for determining the aqueous solubility of organic compounds that was initially developed by May et al. (as described in the references listed in paragraphs (e)(5) and (e)(6) of this section), modified by DeVoe et al. (as described in the reference listed in paragraph (e)(1) of this section), and finalized by Wasik et al. (as described in the reference listed in paragraph (e)(11) of this section). The DCCLC technique utilizes a generator column, extractor column and HPLC coupled or interconnected to provide a continuous closed flow system. Saturated aqueous solutions of the test compound are produced by pumping water through the generator column that is packed with a solid support coated with the compound. The compound is extracted from the saturated solution onto an extractor column, then eluted from the extractor column with a solvent/water mixture and subsequently analyzed by HPLC using a variable wavelength ultraviolet (UV) detector operating at a suitable wavelength. Chromatogram peaks are recorded and integrated using a recording integrator. The concentration of the compound in the effluent from the generator column, i.e., the water solubility of the compound, is determined from the mass of the compound (solute) extracted from a measured volume of water (solvent).
(ii) Since the HPLC method is only applicable to compounds that absorb in the UV, an alternate GC method, or any other reliable procedure (which must be approved by OCSPP), can be used for those compounds that do not absorb in the UV. In the GC method the saturated solutions produced in the generator column are extracted using an appropriate organic solvent that is subsequently injected into the GC, or any other suitable analytical device, for analysis of the test compound.
(4) Reference chemicals. Table 1 of this section lists the water solubilities at 25 °C for a number of reference chemicals as obtained from the scientific literature. The data from Wasik et al. (as described in the reference listed in paragraph (e)(11) of this section), Miller et al. and Tewari et al. (as described in the references listed in paragraphs (e)(7) and (e)(10) of this section, respectively) were obtained from the generator column method. The water solubilities data were also obtained from Mackay et al. and Yalkowski et al. (as described in the references listed in paragraphs (e)(4) and (e)(12) of this section, respectively) and other scientists by the conventional shake flask method. These data have been provided primarily so that the generator column method can be calibrated from time to time and to allow the chemical testing laboratory an opportunity to compare its results with those listed in table 1 of this section. The water solubility values at 25 °C reported by Yalkowski et al. are their preferred values and, in general, represent the best available water solubility data at 25 °C. The testing laboratory has the option of choosing its own reference chemicals, but references must be given to establish the validity of the measured values of the water solubility.
Table 1 - Water Solubilities at 25 °C of Some Reference Chemicals
| Reference chemical | Water solubility (ppm at 25 °C) | ||
|---|---|---|---|
| Wasik (generator column method) | Yalkowski 1 5 | Other literature references | |
| 2-Heptanone | 24080 | 4300 | 54330 |
| 1-Chlorobutane | 2873 | 872.9 | 7666 |
| Ethylbenzene | 2187 | 208 | 7162 |
| 1,2,3-Trimethylbenzene | 265.5 | 75.2 | 748.2 |
| Biphenyl | 3 106.71 | 7.48 | 86.62 |
| Phenanthrene | 41.002 | 1.212 | - |
| 2,4,6-Trichlorobiphenyl | 3 100.226 | 0.225 | 80.119 |
| 2,3,4,5-Tetrachlorobiphenyl | 3 100.0209 | 0.01396 | 80.0192 |
| Hexachlorobenzene | - | 0.004669 | 90.00996 |
| 2,3,4,5,6-Pentachlorobiphenyl | 3 100.00548 | 0.004016 | 80.0068 |
1 Preferred water solubility at 25 °C by Yalkowski et al. (1990) in paragraph (e)(12) of this section based on a critical review of all the experimental water solubility data published.
2 Tewari et al. (1982) in paragraph (e)(10) of this section.
3 Leifer et al. (1983) in paragraph (e)(3) of this section.
4 May, Wasik, and Freeman (1978, 1978a) in paragraphs (e)(5) and (6) of this section.
5 Yalkowski et al. (1990) in paragraph (e)(12) of this section.
6 Hansch et al. (1968) in paragraph (e)(2) of this section.
7 Sutton and Calder (1975) in paragraph (e)(9) of this section.
8 Mackay et al. (1980) in paragraph (e)(4) of this section.
9 The elution chromatographic method from Organization for Economic Cooperation and Development (OECD) (1981) in paragraph (e)(8) of this section.
10 Miller et al. (1984) in paragraph (e)(7) of this section.
(5) Applicability and specificity. (i) Procedures are described in this section to determine the water solubility for liquid or solid compounds. The water solubility can be determined in very pure water, buffer solution for compounds that reversibly ionize or protonate, or in artificial seawater as a function of temperature (i.e., in the range of temperatures of environmental concern). This section is not applicable to the water solubility of gases.
(ii) This section is designed to determine the water solubility of a solid or liquid test chemical in the range of 1 ppb to 5,000 ppm. For chemicals whose solubility is below 1 ppb, the water solubility should be characterized as “less than 1 ppb” with no further quantification. For solubilities greater than 5,000 ppm, the shake flask method should be used, see paragraph (e)(15) of this section.
(c) Test procedure - (1) Test conditions - (i) Special laboratory equipment - (A) Generator column. (1) Either of two different designs shall be used depending on whether the eluted aqueous phase is analyzed by HPLC in paragraph (c)(3)(ii) of this section or by solvent extraction followed by GC (or any other reliable quantitative) analysis of solvent extract in paragraph (c)(3)(iv) of this section. The design of the generator column is shown in the following figure 1:
Figure 1 - Generator Column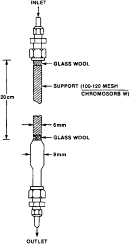
(2) The column consists of a 6 mm ( 1/4 in) O.D. pyrex tube joined to a short enlarged section of 9 mm pyrex tubing which in turn is connected to another section of 6 mm ( 1/4 in) O.D. pyrex tubing. Connections to the inlet teflon tubing ( 1/8 in O.D.) and to the outlet stainless steel tubing ( 1/16 in O.D.) shall be made by means of stainless steel fittings with teflon ferrules. The column is enclosed in a water jacket for temperature control as shown in the following figure 2:
Figure 2 - Setup Showing Generator Column Enclosed in a Water Jacket and Overall Arrangement of the Apparatus Used in the GC Method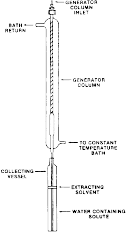
(B) Constant temperature bath with circulation pump-bath and capable of controlling temperature to ±0.05 °C, see paragraph (c)(3) of this section.
(C) HPLC equipped with a variable wavelenth UV absorption detector operating at a suitable wavelength and a recording integrator in paragraph (c)(3)(ii) of this section.
(D) Extractor column - 6.6 × 0.6 cm stainless steel tube with end fittings containing 5 µm frits filled with a superficially porous phase packing (Bondapack C18/Corasil: Waters Associates) in paragraph (c)(3)(ii) of this section.
(E) Two 6-port high-pressure rotary switching valves in paragraph (c)(3)(ii) of this section.
(F) Collection vessel - 8 × 3/4 in section of pyrex tubing with a flat bottom connected to a short section of 3/8 in O.D. borosilicate glass tubing in figure 2 in paragraph (c)(1)(i)(A)(2) of this section. The collecting vessel is sealed with a 3/8 in teflon cap fitting in paragraph (c)(3)(iii) of this section.
(G) GC, or any other reliable analytical equipment, which has a detector sensitive to the solute of interest in paragraph (c)(3)(iii) of this section.
(ii) Purity of water. Water meeting appropriate American Society for Testing and Materials (ASTM) Type II standards, or an equivalent grade, are recommended to minimize the effects of dissolved salts and other impurities on water solubility. ASTM Type II water is presented in the reference listed in paragraph (e)(13) of this section.
(iii) Purity of solvents. All solvents used in this method must be reagent or HPLC grade. Solvents must contain no impurities which could interfere with the determination of the test compound.
(iv) Seawater. When the water solubility in seawater is desired, the artificial seawater described in paragraph (c)(2)(ii) of this section must be used.
(v) Effect of pH on solubility. For chemicals that reversibly ionize or protonate with a pKa or pKb between 3 and 11, experiments must be performed at pH's 5.0, 7.0, and 9.0 using appropriate buffers.
(2) Preparation of reagents and solutions - (i) Buffer solutions. Prepare buffer solutions as follows:
(A) pH 3.0 - to 250 mL of 0.10M potassium hydrogen phosphate add 111 mL of 0.10 M hydrochloric acid; adjust the final volume to 500 mL with reagent grade water.
(B) pH 5.0 - to 250 mL of 0.1M potassium hydrogen phthalate add 113 mL of 0.1M sodium hydroxide; adjust the final volume to 500 mL with reagent grade water.
(C) pH 7.0 - to 250 mL of 0.1M potassium dihydrogen phosphate add 145 mL of 0.1M sodium hydroxide; adjust the final volume to 500 mL with reagent grade water.
(D) pH 9.0 - to 250 mL of 0.075M borax add 69 mL of 0.1M HCl; adjust the final volume to 500 mL with reagent grade water.
(E) pH 11.0 - to 250 mL of 0.05 M sodium bicarbonate add 3 mL of 0.10 M sodium hydroxide; adjust the final volume to 500 mL with reagent grade water.
(ii) Check the pH of each buffer solution with a pH meter at 25 °C and adjust to pH 5.0, 7.0, or 9.0, if necessary. If the pH of the solution has changed by ±0.2 pH units or more after the addition of the test compound, then a more concentrated buffer is required for that pH determination. The sponsor should then choose a more suitable buffer.
(iii) Artificial seawater. Add the reagent-grade chemicals listed in table 2 of this section in the specified amounts and order to 890 mL of reagent-grade water. Each chemical shall be dissolved before another one is added.
Table 2 - Constituents of Artificial Seawater 1
| Chemical | Amount |
|---|---|
| NaF | 3 mg |
| SrCl2,6H2O | 20 mg |
| H3BO3 | 30 mg |
| KBr | 100 mg |
| KCl | 700 mg |
| CaCl2.2H2O | 1.47 gram (g) |
| Na2SO4 | 4.00 g |
| MgCl2.6H2O | 10.78 g |
| NaCl | 23.50 g |
| Na2SiO3.9H2O | 20 mg |
| NaHCO3 | 200 mg |
1 If the resulting solution is diluted to 1 L, the salinity should be 34 ±0.5 g/kilogram (kg) and the pH 8.0 ±0.2. The desired test salinity is attained by dilution at time of use.
(3) Performance of the test. Using either the procedures in paragraph (c)(3)(ii) or (c)(3)(iii) of this section, determine the water solubility of the test compound at 25 °C in reagent-grade water or buffer solution, as appropriate. Under certain circumstances, it may be necessary to determine the water solubility of a test compound at 25 °C in artificial seawater. The water solubility can also be determined at other temperatures of environmental concern by adjusting the temperature of the water bath to the appropriate temperature.
(i) Prior to the determination of the water solubility of the test chemical, two procedures shall be followed.
(A) The saturated aqueous solution leaving the generator column must be tested for the presence of an emulsion, using a Tyndall procedure. If colloids are present, they must be eliminated prior to the injection into the extractor column. This may be achieved by lowering the flow rate of the water.
(B) The efficiency of the removal of the solute (i.e. test chemical) by the solvent extraction from the extraction column must be determined and used in the determination of the water solubility of the test chemical.
(ii) Procedure A - HPLC method - (A) Scope. (1) Procedure A covers the determination of the aqueous solubility of compounds which absorb in the UV.
(i) The HPLC analytical system is shown schematically in the following figure 3:
Figure 3 - Schematic of HPLC - Generator Column Flow System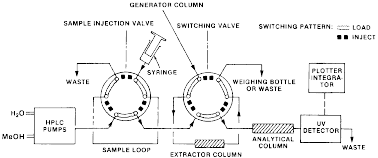
(ii) Two reciprocating piston pumps deliver the mobile phase (water or solvent/water mixture) through two 6-port high-pressure rotary valves and a 30 × 0.6 cm C18/Corasil analytical column to a variable wavelength UV absorption detector operating at a suitable wavelength; chromatogram peaks are recorded and integrated with a recording integrator. One of the 6-port valves is the sample injection valve used for injecting samples of standard solutions of the solute in an appropriate concentration for determining RFs of standard solutions of basic chromate for determining the sample-loop volume. The other 6-port valve in the system serves as a switching valve for the extractor column which is used to remove solute from the aqueous solutions.
(2) The general procedure for analyzing the aqueous phase is as follows (a detailed procedure is given in paragraph (c)(3)(ii)(B)(4) of this section).
(i) Direct the aqueous solution to “Waste,” see figure 3 in paragraph (c)(3)(ii)(A)(1)(i) of this section, with the switching valve in the inject position in order to equilibrate internal surfaces with the solution, thus ensuring that the analyzed sample would not be depleted by solute adsorption on surfaces upstream from the valve.
(ii) At the same time, water is pumped from the HPLC pumps in order to displace the solvent from the extractor column.
(iii) The switching valve is next changed to the load position to divert a sample of the solution through the extractor column, and the liquid leaving this column is collected in a weighing bottle. During this extraction step, the mobile phase is changed to a solvent/water mixture to condition the analytical column.
(iv) After the desired volume of sample is extracted, the switching valve is returned to the inject position for elution and analysis. Assuming that there is no breakthrough of solute from the extractor column during the extraction step, the chromatographic peak represents all of the solute in the sample, provided that the extraction efficiency is 100%. If the extraction efficiency is less than 100%, then the extraction efficiency shall be used to determine the actual weight of the solute extracted.
(v) The solute concentration in the aqueous phase is calculated from the peak area and the weight of the extracted liquid collected in the weighing bottle.
(B) Determinations - (1) Sample-loop volume. Accurate measurement of the sample loop may be accomplished by using the spectrophotometric method of Devoe et al. under paragraph (e)(1) of this section. For this method measure absorbance, Aloop, at 373 nm of at least three solutions, each of which is prepared by collecting from the sample valve an appropriate number, n, of loopfuls of an aqueous stock solution of K2CrO4 (1.3% by weight) and diluting to 50 mL with 0.2% KOH. (For a 20 µL loop, use n = 5; for a 50 µL loop, use n = 2.) Also measure the absorbance, Astock, of the same stock solution after diluting 1:500 with 0.2% KOH. Calculate the loop volume to the nearest 0.1 µL using the equation:
(2) RF. (i) For all determinations adjust the mobile phase solvent/water ratio and flow rate to obtain a reasonable retention time on the HPLC column. For example, typical concentrations of solvent in the mobile phase range from 50 to 100% while flow rates range from 1 to 3 mL/min; these conditions give a 3 to 5 min retention time.
(ii) Prepare standard solutions of known concentrations of the solute in a suitable solvent. Concentrations must give a recorder response within the maximum response of the detector. Inject samples of each standard solution into the HPLC system using the calibrated sample loop. Obtain an average peak area from at least three injections of each standard sample at a set absorbance unit full scale (AUFS), i.e., at the same absorbance scale attenuation setting.
(iii) Calculate the RF from the following equation:
(3) Loading of the generator column. (i) The design of the generator column was described in paragraph (c)(1)(i) of this section and is shown in figure 1 in paragraph (c)(1)(i)(A) of this section. To pack the column, a plug of silanized glass wool is inserted into one end of the 6 mm pyrex tubing. Silanized diatomaceous silica support (about 0.5g 100-120 mesh Chromosorb (W) chromatographic support material) is poured into the tube with tapping and retained with a second plug of silanized glass wool.
(ii) If the solute is a liquid, the column is loaded by pulling the liquid solute through the dry support with gentle suction. If the solute is a solid, a 1% solution of the solid in a volatile solvent is added to the dry packing. The solvent is then distilled off the column under reduced pressure. After loading the column draw water up through the column to remove entrapped air.
(4) Analysis of the solute. Use the following procedure to collect and analyze the solute.
(i) With the switching valve (figure 3 in paragraph (c)(3)(ii)(A)(1)(i) of this section) in the inject position (i.e., water to waste), pump water through the generator column at a flow rate of approximately 1 mL/min for approximately 5 minutes (min) to bring the system into equilibrium. Pump water to the generator column by means of a minipump or pressurized water reservoir as shown in the following figure 4:
Figure 4 - Water Reservoir for GC Method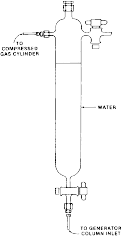
(ii) Flush out the solvent that remains in the system from previous runs by changing the mobile phase to 100% H2O and allowing the water to reach the HPLC detector, as indicated by a negative reading. As soon as this occurs, place a 25 mL weighing bottle (weighed to the nearest mg) at the waste position and immediately turn the switching valve to the load position.
(iii) Collect an amount of water (as determined by trial and error) in the weighing bottle, corresponding to the amount of solute adsorbed by the extractor column that gives a large on-scale detector response. During this extraction step, switch back to the original HPLC mobile phase composition, i.e., solvent/water mixture, to condition the HPLC analytical column.
(iv) After the desired volume of sample has been extracted, turn the switching valve back to the inject position (figure 3 in paragraph (c)(3)(ii)(A)(1)(i) of this section); at the same time turn on the recording integrator. The solvent/water mobile phase will elute the solute from the extractor column and transfer the solute to the HPLC analytical column.
(v) Remove the weighing bottle, cap it, and replace it with the waste container. Determine the weight of water collected to the nearest mg and record the corresponding peak area. Using the same AUFS setting repeat the analysis of the solute at least two more times and determine the average ratio of peak area to grams of water collected. In this equation, s = solubility (M), RF = response factor, Vloop = sample-loop volume (L), and R = ratio of area to grams of water. Calculate the solute solubility in water using the following equation:
(iii) Procedure B - GC method - (A) Scope. In the GC method, or any other analytical method, aqueous solutions from the generator column enter a collecting vessel (figure 2 in paragraph (c)(1)(i)(A)(2) of this section) containing a known weight of extracting solvent which is immiscible in water. The outlet of the generator column is positioned such that the aqueous phase always enters below the extracting solvent. After the aqueous phase is collected, the collecting vessel is stoppered and the quantity of aqueous phase is determined by weighing. The solvent and the aqueous phase are equilibrated by slowly rotating the collecting vessel. The extraction efficiency of the solvent must be determined at this time. A small amount of the extracting solvent is removed and injected into a gas chromograph equipped with an appropriate detector. The solute concentration in the aqueous phase is determined from a calibration curve constructed using known concentrations of the solute.
(B) Alternative method. If another (approved) analytical method is used instead of the GC, that method shall be used to determine quantitatively the amount of solute present in the extraction solvent.
(C) Determinations - (1) Calibration curve. (i) Prepare solute standard solutions of concentrations covering the range of the solute solubility. Select a column and optimum GC operating conditions for resolution between the solute and solvent and the solute and extracting solvent. Inject a known volume of each standard solution into the injection port of the GC. For each standard solution determine the average of the ratio R of peak area to volume (in microliters) for three chromatographic peaks from three injections.
(ii) After running all the standard solutions, determine the coefficients, a and b, using a linear regression equation of C vs. R in the following form:
(iii) If another analytical method is used, the procedures described in paragraph (c)(3)(iii)(C)(1) of this section shall be used to determine quantitatively the amount of solute in the extraction solvent.
(2) Loading of the generator column. The generator column is packed and loaded with solute in the same manner as for the HPLC method described under paragraph (c)(3)(ii)(B)(3) of this section. As shown in figure 2 in paragraph (c)(1)(i)(A)(2) of this section, attach approximately 20 cm of straight stainless steel tubing to the bottom of the generator column. Connect the top of the generator column to a water reservoir (figure 4 in paragraph (c)(3)(ii)(B)(4)(i) of this section) using teflon tubing. Use air or nitrogen pressure (5 PSI) from an air or nitrogen cylinder to force water from the reservoir through the column. Collect water in an Erlenmeyer flask for approximately 15 min while the solute concentration in water equilibrates; longer time may be required for less soluble compounds.
(3) Collection and extraction of the solute. During the equilibration time, add a known weight of extracting solvent to a collection vessel which can be capped. The extracting solvent should cover the bottom of the collection vessel to a depth sufficient to submerge the collecting tube but still maintain 100:1 water/solvent ratio. Record the weight (to the nearest mg) of a collection vessel with cap and extracting solvent. Place the collection vessel under the generator column so that water from the collecting tube enters below the level of the extracting solvent (figure 2 in paragraph (c)(1)(i)(A)(2) of this section). When the collection vessel is filled, remove it from under the generator column, replace cap, and weigh the filled vessel. Determine the weight of water collected. Before analyzing for the solute, gently shake the collection vessel contents for approximately 30 min, controlling the rate of shaking so as not to form an emulsion; rotating the flask end over end five times per minute is sufficient.
(4) Analysis of the solute. (i) After shaking, allow the collection vessel to stand for approximately 30 min; then remove a known volume of the extracting solvent from the vessel using a microliter syringe and inject it into the GC. Record the ratio of peak area to volume injected and, from the regression equation of the calibration line, determine the concentration of solute in the extracting solvent. In this equation, Ces is the concentration of solute in extracting solvent (M), dH2O and des are the densities of water and extracting solvent, respectively, and ges and gH2O are the grams of extracting solvent and water, respectively, contained in the collection vessel. The concentration of solute in water C(M) is determined from the following equation:
(ii) Make replicate injections from each collecting vessel to determine the average solute concentration in water for each vessel. To make sure the generator column has reached equilibrium, run at least two additional (for a total of three) collection vessels and analyze the extracted solute as described above. Calculate the water solubility of the solute from the average solute concentration in the three vessels.
(iv) Modification of procedures for potential problems. If the test compound decomposes in one or more of the aqueous solvents required during the period of the test at a rate such that an accurate value for water solubility cannot be obtained, then it will be necessary to carry out detailed transformation studies; e.g., hydrolysis in paragraph (e)(16) of this section. If decomposition is due to aqueous photolysis, then it will be necessary to carry out water solubility studies in the dark, under red or yellow lights, or by any other suitable method to eliminate this transformation process.
(d) Data and reporting - (1) Test report. (i) For each set of conditions, (e.g., temperature, pure water, buffer solution, artificial seawater) required for the study, provide the water solubility value for each of three determinations, the mean value, and the standard deviation.
(ii) For compounds that decompose at a rate such that a precise value for the water solubility cannot be obtained, provide a statement to that effect.
(iii) For compounds with water solubility below 1 ppb, report the value as “less than 1 ppb.”
(2) Specific analytical, calibration, and recovery procedures. (i) For the HPLC method describe and/or report:
(A) The method used to determine the sample-loop volume and the average and standard deviation of that volume.
(B) The average and standard deviation of the RF.
(C) Any changes made or problems encountered in the test procedure.
(ii) For the GC, or any other analytical, method report:
(A) The column and GC operating conditions of temperature and flow rate, or the operating conditions of any other analytical method used.
(B) The average and standard deviation of the average area per microliter obtained for each of the standard solutions.
(C) The form of the regression equation obtained in the calibration procedure.
(D) The extracting solvent used, and its extraction efficiency.
(E) The average and standard deviation of solute concentration in each collection vessel.
(F) Any changes made or problems encountered in the test procedure.
(G) If applicable, a complete description of the analytical method which was used instead of the GC method.
(e) References. For additional information on this test guideline, the following references should be consulted. These references are available at the addresses in § 700.17(b)(1) and (2) of this chapter.
(1) DeVoe, H. et al., Generator columns and high pressure liquid chromatography for determining aqueous solubilities and octanol-water partition coefficients of hydrophobic substances. Journal of Research, National Bureau of Standards, 86:361-366 (1981).
(2) Hansch, C. et al., The linear free-energy relationship between partition coefficients, and the aqueous solubility of organic liquids. Journal of Organic Chemistry 33:347-350 (1968).
(3) Leifer, A. et al., Environmental transport and transformation of polychlorinated biphenyls. Chapter 1. U.S. Environmental Protection Agency Report: EPA-560/5-83-005 (1983).
(4) Mackay, D. et al., Relationships between aqueous solubility and octanol-water partition coefficient. Chemosphere 9:701-711 (1980).
(5) May, W.E. et al., Determination of the aqueous solubility of polynuclear aromatic hydrocarbons by a coupled column liquid chromatographic technique. Analytical Chemistry 50:175-179 (1978).
(6) May, W.E. et al. Determination of the solubility behavior of some polycyclic aromatic hydrocarbons in the water. Analytical Chemistry, 50:997-1000 (1978a).
(7) Miller, N.M. et al., Aqueous solubilities, octanol/water partition coefficients, and entropy of melting of chlorinated benzenes and biphenyls. Journal of Chemical and Engineering Data 29:184-190 (1984).
(8) OECD/Organization for Economic Cooperation and Development. Test Guideline No. 105. Water solubility column elution-flask method (1981).
(9) Sutton, C. and Calder, J.A., Solubility of alkylbenzenes in distilled water and seawater at 25 °C. Journal of Chemical and Engineering Data 20:320-322 (1975).
(10) Tewari, Y.B. et al., Aqueous solubility and octanol/water partition coefficient of organic compounds at 25 °C. Journal of Chemical and Engineering Data 27:451-454 (1982).
(11) Wasik, S.P. et al., Octanol/Water Partition Coefficient and Aqueous Solubilities of Organic Compounds. NBS Report NBSIR 81-2406. Washington, DC: National Bureau of Standards, U.S. Department of Commerce (1981).
(12) Yalkowski, S.H. et al., “Aquasol database of aqueous solubilities of organic compounds”; Fifth Edition. University of Arizona, College of Pharmacy, Tucson, AZ 85721 (1990) (available at http://www.pharm.arizona.edu/aquasol/index.html).
(13) ASTM D 1193-91, Standard Specification for Reagent Water. American Society for Testing and Materials (ASTM). 1916 Race St., Philadelphia, PA 19103.
[65 FR 78751, Dec. 15, 2000, as amended at 77 FR 46293, Aug. 3, 2012]