Title 40
SECTION 796.1950
796.1950 Vapor pressure.
§ 796.1950 Vapor pressure.(a) Introduction - (1) Background and purpose. (i) Volatilization, the evaporative loss of a chemical, depends upon the vapor pressure of chemical and on environmental conditions which influence diffusion from a surface. Volatilization is an important source of material for airborne transport and may lead to the distribution of a chemical over wide areas and into bodies of water far from the site of release. Vapor pressure values provide indications of the tendency of pure substances to vaporize in an unperturbed situation, and thus provide a method for ranking the relative volatilities of chemicals. Vapor pressure data combined with water solubility data permit the calculation of Henry's law constant, a parameter essential to the calculation of volatility from water.
(ii) Chemicals with relatively low vapor pressures, high adsorptivity onto solids, or high solubility in water are less likely to vaporize and become airborne than chemicals with high vapor pressures or with low water solubility or low adsorptivity to solids and sediments. In addition, chemicals that are likely to be gases at ambient temperatures and which have low water solubility and low adsorptive tendencies are less likely to transport and persist in soils and water. Such chemicals are less likely to biodegrade or hydrolyze and are prime candidates for atmospheric oxidation and photolysis (e.g., smog formation or stratospheric alterations). On the other hand, nonvolatile chemicals are less frequently involved in atmosphere transport, so that concerns regarding them should focus on soils and water.
(iii) Vapor pressure data are an important consideration in the design of other chemical fate and effects tests; for example, in preventing or accounting for the loss of violatile chemicals during the course of the test.
(2) Definitions and units. (i) “Desorption efficiency” of a particular compound applied to a sorbent and subsequently extracted with a solvent is the weight of the compound which can be recovered from the sorbent divided by the weight of the compound originally sorbed.
(ii) “Pascal” (Pa) is the standard international unit of vapor pressure and is defined as newtons per square meter (N/m 2). A newton is the force necessary to give acceleration of one meter per second squared to one kilogram of mass.
(iii) The “torr” is a unit of pressure which equals 133.3 pascals or 1 mm Hg at 0 °C.
(iv) “Vapor pressure” is the pressure at which a liquid or solid is in equilibrium with its vapor at a given temperature.
(v) “Volatilization” is the loss of a substance to the air from a surface or from solution by evaporation.
(3) Principle of the test methods. (i) The isoteniscope procedure uses a standardized technique [ASTM 1978] that was developed to measure the vapor pressure of certain liquid hydrocarbons. The sample is purified within the equipment by removing dissolved and entrained gases until the measured vapor pressure is constant, a process called “degassing.” Impurities more volatile than the sample will tend to increase the observed vapor pressure and thus must be minimized or removed. Results are subject to only slight error for samples containing nonvolatile impurities.
(ii) Gas saturation (or transpiration) procedures use a current of inert gas passed through or over the test material slowly enough to ensure saturation and subsequent analysis of either the loss of material or the amount (and sometimes kind) of vapor generated. Gas saturation procedures have been described by Spencer and Cliath (1969) under paragraph (d)(2) of this section. Results are easy to obtain and can be quite precise. The same procedures also can be used to study volatilization from laboratory scale environmental simulations. Vapor pressure is computed on the assumption that the total pressure of a mixture of gases is equal to the sum of the pressures of the separate or component gases and that the ideal gas law is obeyed. The partial pressure of the vapor under study can be calculated from the total gas volume and the weight of the material vaporized. If v is the volume which contains w grams of the vaporized material having a molecular weight M, and if p is the pressure of the vapor in equilibrium at temperature T (K), then the vapor pressure, p, of the sample is calculated by
p = (w/M)(RT/v), where R is the gas constant (8.31 Pa m 2 mol−1 K−1) when the pressure is in pascals (Pa) and the volume is in cubic meters. As noted by Spencer and Cliath (1970) under paragraph (d)(3) of this section, direct vapor pressure measurements by gas saturation techniques are more directly related to the volatilization of chemicals than are other techniques.(iii) In an effort to improve upon the procedure described by Spencer and Cliath (1969) under paragraph (d)(2) of this section, and to determine the applicability of the gas saturation method to a wide variety of chemical types and structures, EPA has sponsored research and development work at SRI International (EPA 1982) under paragraph (d)(1) of this section. The procedures described in this Test Guideline are those developed under that contract and have been evaluated with a wide variety of chemicals of differing structure and vapor pressures.
(4) Applicability and specificity. (i) A procedure for measuring the vapor pressure of materials released to the environment ideally would cover a wide range of vapor pressure values, at ambient temperatures. No single procedure can cover this range, so two different procedures are described in this section, each suited for a different part of the range. The isoteniscope procedure is for pure liquids with vapor pressures from 0.1 to 100 kPa. For vapor pressures of 10−5 to 10 3 Pa, a gas saturation procedure is to be used.
(ii) With respect to the isoteniscope method, if compounds that boil close to or form azeotropes with the test material are present, it is necessary to remove the interfering compounds and use pure test material. Impurities more volatile than the sample will tend to increase the observed vapor pressure above its true value but the purification steps will tend to remove these impurities. Soluble, nonvolatile impurities will decrease the apparent vapor pressure. However, because the isoteniscope procedure is a static, fixed-volume method in which an insignificant fraction of the liquid sample is vaporized, it is subject to only slight error for samples containing nonvolatile impurities. That is, the nonvolatile impurities will not be concentrated due to vaporization of the sample.
(iii) The gas saturation method is applicable to solid or liquid chemicals. Since the vapor pressure measurements are made at ambient temperatures, the need to extrapolate data from high temperatures is not necessary and high temperature extrapolation, which can often cause serious errors, is avoided. The method is most reliable for vapor pressures below 10 3 Pa. Above this limit, the vapor pressures are generally overestimated, probably due to aerosol formation. Finally, the gas saturation method is applicable to the determination of the vapor pressure of impure materials.
(b) Test procedures - (1) Test conditions. (i) The apparatus in the isoteniscope method is described in paragraph (b)(2)(i) of this section.
(ii) The apparatus used in the gas saturation method is described in paragraph (b)(2)(ii) of this section.
(2) Performance of the tests - (i) Isoteniscope Procedure. The isoteniscope procedure described as ANSI/ASTM Method D 2879-86 is applicable for the measurement of vapor pressures of liquids with vapor pressures of 0.1 to 100 kilopascals (kPa) (0.75 to 750 torr). ASTM D 2879-86 is available for inspection at the National Archives and Records Administration (NARA). For information on the availability of this material at NARA, call 202-741-6030, or go to: http://www.archives.gov/federal_register/code_of_federal_regulations/ibr_locations.html.\n. This incorporation by reference was approved by the Director of the Office of the Federal Register. This material is incorporated as it exists on the date of approval and a notice of any change in this material will be published in the Federal Register. Copies of the incorporated material may be obtained from the Director, Environmental Assistance Division (7408), Office of Pollution Prevention and Toxics, Environmental Protection Agency, Room E-543B, 1200 Pennsylvania Ave. NW., Washington, DC 20460-0001, or from the American Society for Testing and Materials (ASTM), 1916 Race Street, Philadelphia, PA 19103. The isoteniscope method involves placing liquid sample in a thermostated bulb (the isoteniscope) connected to a manometer and a vacuum pump. Dissolved and entrained gases are removed from the sample in the isoteniscope by degassing the sample at reduced pressure. The vapor pressure of the sample at selected temperatures is determined by balancing the pressure due to the vapor of the sample against a known pressure of an inert gas. The vapor pressure of the test compound is determined in triplicate at 25 ±0.5 °C and at any other suitable temperatures (±0.5°). It is important that additional vapor pressure measurements be made at other temperatures, as necessary, to assure that there is no need for further degassing, as described in the ASTM method.
(ii) Gas saturation procedure. (A) The test procedures require the use of a constant-temperature box as depicted in the following Figure 1.
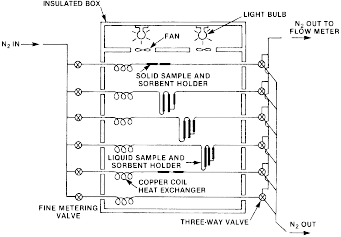 Figure 1
- Schematic Diagram of Vapor Saturation Apparatus The insulated
box, containing sample holders, may be of any suitable size and
shape. The sketch in Figure 1 shows a box containing three solid
sample holders and three liquid sample holders, which allows for
the triplicate analysis of either a solid or liquid sample. The
temperature within the box is controlled to ±0.5° or better.
Nitrogen gas, split into six streams and controlled by fine needle
valves (approximately 0.79 mm orifice), flows into the box via 3.8
mm (0.125 in.) i.d. copper tubing. After temperature equilibration,
the gas flows through the sample and the sorbent trap and exits
from the box. The flow rate of the effluent carrier gas is measured
at room temperature with a bubble flow meter or other suitable
device. The flow rate is checked frequently during the experiment
to assure that there is an accurate value for the total volume of
carrier gas. The flow rate is used to calculate the total volume
(at room temperature) of gas that has passed through the sample and
sorbent [(vol/time) × time = volume]. The vapor pressure of the
test substance can be calculated from the total gas volume and the
mass of sample vaporized. If v is the volume of gas that
transported mass w of the vaporized test material having a
molecular weight M, and if p is the equilibrium vapor pressure of
the sample at temperature T, then p is calculated by the equation p
= (w/M)(RT/v). In this equation, R is the gas constant (8.31 Pa m
3mol−1 K−1). The pressure is expressed in pascals (Pa), the volume
in cubic meters (m 3), mass in grams and T in kelvins (K). T =
273.15 + t, if t is measured in degrees Celsius (°C).
Figure 1
- Schematic Diagram of Vapor Saturation Apparatus The insulated
box, containing sample holders, may be of any suitable size and
shape. The sketch in Figure 1 shows a box containing three solid
sample holders and three liquid sample holders, which allows for
the triplicate analysis of either a solid or liquid sample. The
temperature within the box is controlled to ±0.5° or better.
Nitrogen gas, split into six streams and controlled by fine needle
valves (approximately 0.79 mm orifice), flows into the box via 3.8
mm (0.125 in.) i.d. copper tubing. After temperature equilibration,
the gas flows through the sample and the sorbent trap and exits
from the box. The flow rate of the effluent carrier gas is measured
at room temperature with a bubble flow meter or other suitable
device. The flow rate is checked frequently during the experiment
to assure that there is an accurate value for the total volume of
carrier gas. The flow rate is used to calculate the total volume
(at room temperature) of gas that has passed through the sample and
sorbent [(vol/time) × time = volume]. The vapor pressure of the
test substance can be calculated from the total gas volume and the
mass of sample vaporized. If v is the volume of gas that
transported mass w of the vaporized test material having a
molecular weight M, and if p is the equilibrium vapor pressure of
the sample at temperature T, then p is calculated by the equation p
= (w/M)(RT/v). In this equation, R is the gas constant (8.31 Pa m
3mol−1 K−1). The pressure is expressed in pascals (Pa), the volume
in cubic meters (m 3), mass in grams and T in kelvins (K). T =
273.15 + t, if t is measured in degrees Celsius (°C).
(B) Solid samples are loaded into 5 mm i.d. glass tubing between glass wool plugs. The following Figure 2 depicts a drawing of a sample holder and absorber system.
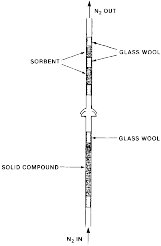 Figure 2
- Solid Compound Sampling System
Figure 2
- Solid Compound Sampling System
(C) Liquid samples are contained in a holder as shown in the following Figure 3.
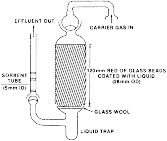 Figure 3
- Liquid Compound Sampling System The most reproducible method for
measuring the vapor pressure of liquids is to coat the liquid on
glass beads and to pack the holder in the designated place with
these beads.
Figure 3
- Liquid Compound Sampling System The most reproducible method for
measuring the vapor pressure of liquids is to coat the liquid on
glass beads and to pack the holder in the designated place with
these beads.
(D) At very low vapor pressures and sorbent loadings, adsorption of the chemical on the glass wool separating the sample and the sorbent and on the glass surfaces may be a serious problem. Therefore, very low loadings should be avoided whenever possible. Incoming nitrogen gas (containing no interfering impurities) passes through a coarse frit and bubbles through a 38 cm column of liquid sample. The stream passes through a glass wool column to trap aerosols and then through a sorbent tube, as described above. The pressure drop across the glass wool column and the sorbent tube are negligible.
(E) With both solid and liquid samples, at the end of the sampling time, the front and backup sorbent sections are analyzed separately. The compound on each section is desorbed by adding the sorbent from that section to 1.0 ml of desorption solvent in a small vial and allowing the mixture to stand at a suitable temperature until no more test compound desorbs. It is extremely important that the desorption solvent contain no impurities which would interfere with the analytical method of choice. The resulting solutions are analyzed quantitatively by a suitable analytical method to determine the weight of sample desorbed from each section. The choice of the analytical method, sorbent, and desorption solvent is dictated by the nature of the test material. Commonly used sorbents include charcoal, Tenax GC, and XAD-2. Describe in detail the sorbent, desorption solvent, and analytical methods employed.
(F) Measure the desorption efficiency for every combination of sample, sorbent, and solvent used. The desorption efficiency is determined by injecting a known mass of sample onto a sorbent and later desorbing it and analyzing for the mass recovered. For each combination of sample, sorbent, and solvent used, carry out the determination in triplicate at each of three concentrations. Desorption efficiency may vary with the concentration of the actual sample and it is important to measure the efficiency at or near the concentration of sample under gas saturation test procedure conditions.
(G) To assure that the gas is indeed saturated with test compound vapor, sample each compound at three differing gas flow rates. Appropriate flow rates will depend on the test compound and test temperature. If the calculated vapor pressure shows no dependence on flow rate, then the gas is assumed to be saturated.
(c) Data and reporting. (1) Report the triplicate calculated vapor pressures for the test material at each temperature, the average calculated vapor pressure at each temperature, and the standard deviation.
(2) Provide a description of analytical methods used to analyze for the test material and all analytical results.
(3) For the isoteniscope procedure, include the plot of p vs. the reciprocal of the temperature in K, developed during the degasing step and showing linearity in the region of 298.15 K (25 °C) and any other required test temperatures.
(4) For the gas saturation procedure, include the data on the calculation of vapor pressure at three or more gas flow rates at each test temperature, showing no dependence on flow rate. Include a description of sorbents and solvents employed and the desorption efficiency calculations.
(5) Provide a description of any difficulties experienced or any other pertinent information.
(d) References. For additional background information on this test guideline the following references should be consulted:
(1) U.S. Environmental Protection Agency. Evaluation of Gas Saturation Methods to Measure Vapor Pressures: Final Report, EPA Contract No. 68-01-5117 with SRI International, Menlo Park, California (1982).
(2) Spencer, W.F. and Cliath, M.M. “Vapor Density of Dieldrin,” Journal of Agricultural and Food Chemistry, 3:664-670 (1969).
(3) Spencer, W.F. and Cliath, M.M. “Vapor Density and Apparent Vapor Pressure of Lindane,” Journal of Agricultural and Food Chemistry, 18:529-530 (1970).
[50 FR 39252, Sept. 27, 1985, as amended at 53 FR 12525, Apr. 15, 1988; 53 FR 21641, June 9, 1988; 60 FR 34466, July 3, 1995; 69 FR 18803, Apr. 9, 2004; 77 FR 46293, Aug. 3, 2012]