Title 40
PART 763 APPENDIX E
| Mineral | Morphology, color a | Refractive indices b | Birefring- ence | Extinction | Sign of elonation | |
|---|---|---|---|---|---|---|
| α | γ | |||||
| Chrysotile (asbestiform serpentine) | Wavy fibers. Fiber bundles have splayed ends and “kinks”. Aspect ratio typically >10:1. Colorless 3, nonpleochroic | 1.493-1.560 | 1.517-1.562 f (normally 1.556) | .008 | | to fiber length | + (length slow) |
| Amosite (asbestiform grunerite) | Straight, rigid fibers. Aspect ratio typically >10:1. Colorless to brown, nonpleochroic or weakly so. Opaque inclusions may be present | 1.635-1.696 | 1.655-1.729 f (normally 1.696-1.710 | .020-.033 | | to fiber length | + (length slow) |
| Crocidolite (asbestiform Riebeckite) | Straight, rigid fibers. Thick fibers and bundles common, blue to purple-blue in color. Pleochroic. Birefringence is generally masked by blue color | 1.654-1.701 | 1.668-1.717 3e (normally close to 1.700) | .014-.016 | | to fiber length | − (length fast) |
| Anthophyllite-asbestos | Straight fibers and acicular cleavage fragments. d Some composite fibers. Aspect ratio <10:1. Colorless to light brown | 1.596-1.652 | 1.615-1.676 f | .019-.024 | | to fiber length | + (length slow) |
| Tremolite-actinolite-asbestos | Normally present as acicular or prismatic cleavage fragments. d Single crystals predominate, aspect ratio <10:1. Colorless to pale green | 1.599-1.668 | 1.622-1.688 f | .023-.020 | Oblique extinction, 10-20° for fragments. Composite fibers show | extinction | + (length slow) |
a From reference 5; colors cited are seen by observation with plane polarized light.
b From references 5 and 8.
c Fibers subjected to heating may be brownish.
d Fibers defined as having aspect ratio >3:1.
e to fiber length.
f |To fiber length.
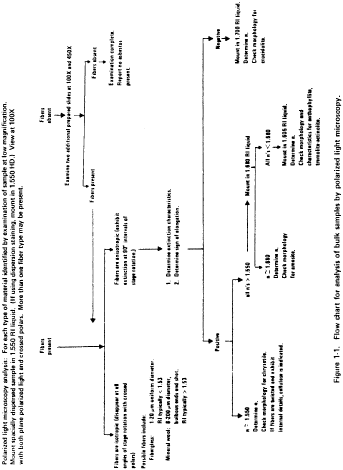
Table 1-2 - Central Stop Dispersion Staining Colors a
| Mineral | RI Liquid | η | η| |
|---|---|---|---|
| Chrysotile | 1.550 HD | Blue | Blue-magenta |
| Amosite | 1.680 | Blue-magenta to pale blue | Golden-yellow |
| 1.550 HD | Yellow to white | Yellow to white | |
| Crocidolite b | 1.700 | Red magenta | Blue-magenta |
| 1.550 HD | Yellow to white | Yellow to white | |
| Anthophyllite | 1.605 HD | Blue | Gold to gold-magenta |
| Tremolite | 1.605 HD c | Pale blue | Gold |
| Actinolite | 1.605 HD | Gold-magenta to blue | Gold |
| 1.630 HD c | Magenta | Golden-yellow |
a From reference 9.
b Blue absorption color.
c Oblique extinction view.
Asbestos quantitation is performed by a point-counting procedure or an equivalent estimation method. An ocular reticle (cross-hair or point array) is used to visually superimpose a point or points on the microscope field of view. Record the number of points positioned directly above each kind of particle or fiber of interest. Score only points directly over asbestos fibers or nonasbestos matrix material. Do not score empty points for the closest particle. If an asbestos fiber and a matrix particle overlap so that a point is superimposed on their visual intersection, a point is scored for both categories. Point counting provides a determination of the area percent asbestos. Reliable conversion of area percent to percent of dry weight is not currently feasible unless the specific gravities and relative volumes of the materials are known.
For the purpose of this method, “asbestos fibers” are defined as having an aspect ratio greater than 3:1 and being positively identified as one of the minerals in Table 1-1.
A total of 400 points superimposed on either asbestos fibers or nonasbestos matrix material must be counted over at least eight different preparations of representative subsamples. Take eight forcep samples and mount each separately with the appropriate refractive index liquid. The preparation should not be heavily loaded. The sample should be uniformly dispersed to avoid overlapping particles and allow 25-50 percent empty area within the fields of view. Count 50 nonempty points on each preparation, using either
• A cross-hair reticle and mechanical stage; or • A reticle with 25 points (Chalkley Point Array) and counting at least 2 randomly selected fields. For samples with mixtures of isotropic and anisotropic materials present, viewing the sample with slightly uncrossed polars or the addition of the compensator plate to the polarized light path will allow simultaneous discrimination of both particle types. Quantitation should be performed at 100X or at the lowest magnification of the polarized light microscope that can effectively distinguish the sample components. Confirmation of the quantitation result by a second analyst on some percentage of analyzed samples should be used as standard quality control procedure.The percent asbestos is calculated as follows:
% asbestos = (a/n) 100% where a = number of asbestos counts, n = number of nonempty points counted (400).If a = 0, report “No asbestos detected.” If 0<a≤3, report “<1% asbestos”.
The value reported should be rounded to the nearest percent.
1.8 References1. Paul F. Kerr, Optical Mineralogy, 4th ed., New York, McGraw-Hill, 1977.
2. E. M. Chamot and C. W. Mason, Handbook of Chemical Microscopy, Volume One, 3rd ed., New York: John Wiley & Sons, 1958.
3. F. Chayes, Petrographic Modal Analysis: An Elementary Statistical Appraisal, New York: John Wiley & Sons, 1956.
4. E. P. Brantly, Jr., K. W. Gold, L. E. Myers, and D. E. Lentzen, Bulk Sample Analysis for Asbestos Content: Evaluation of the Tentative Method, U.S. Environmental Protection Agency, October 1981.
5. U.S. Environmental Protection Agency, Asbestos-Containing Materials in School Buildings: A Guidance Document, Parts 1 and 2, EPA/OPPT No. C00090, March 1979.
6. D. Lucas, T. Hartwell, and A. V. Rao, Asbestos-Containing Materials in School Buildings: Guidance for Asbestos Analytical Programs, EPA 560/13-80-017A, U.S. Environmental Protection Agency, December 1980, 96 pp.
7. D. H. Taylor and J. S. Bloom, Hexametaphosphate pretreatment of insulation samples for identification of fibrous constituents, Microscope, 28, 1980.
8. W. J. Campbell, R. L. Blake, L. L. Brown, E. E. Cather, and J. J. Sjoberg. Selected Silicate Minerals and Their Asbestiform Varieties: Mineralogical Definitions and Identification-Characterization, U.S. Bureau of Mines Information Circular 8751, 1977.
9. Walter C. McCrone, Asbestos Particle Atlas, Ann Arbor: Ann Arbor Science Publishers, June 1980.
Section 2. X-Ray Powder Diffraction 2.1 Principle and ApplicabilityThe principle of X-ray powder diffraction (XRD) analysis is well established. 1 2 Any solid, crystalline material will diffract an impingent beam of parallel, monochromatic X-rays whenever Bragg's Law,
λ = 2d sin θ, is satisfied for a particular set of planes in the crystal lattice, where λ = the X-ray wavelength, Å; d = the interplanar spacing of the set of reflecting lattice planes, Å; and θ = the angle of incidence between the X-ray beam and the reflecting lattice planes. By appropriate orientation of a sample relative to the incident X-ray beam, a diffraction pattern can be generated that, in most cases, will be uniquely characteristic of both the chemical composition and structure of the crystalline phases present.Unlike optical methods of analysis, however, XRD cannot determine crystal morphology. Therefore, in asbestos analysis, XRD does not distinguish between fibrous and nonfibrous forms of the serpentine and amphibole minerals (Table 2-1). However, when used in conjunction with optical methods such as polarized light microscopy (PLM), XRD techniques can provide a reliable analytical method for the identification and characterization of asbestiform minerals in bulk materials.
For qualitative analysis by XRD methods, samples are initially scanned over limited diagnostic peak regions for the serpentine (∼7.4 Å) and amphibole (8.2-8.5 Å) minerals (Table 2-2). Standard slow-scanning methods for bulk sample analysis may be used for materials shown by PLM to contain significant amounts of asbestos (>5-10 percent). Detection of minor or trace amounts of asbestos may require special sample preparation and step-scanning analysis. All samples that exhibit diffraction peaks in the diagnostic regions for asbestiform minerals are submitted to a full (5°-60° 2θ; 1° 2θ/min) qualitative XRD scan, and their diffraction patterns are compared with standard reference powder diffraction patterns 3 to verify initial peak assignments and to identify possible matrix interferences when subsequent quantitative analysis will be performed.
Table 2-1 - The Asbestos Minerals and Their Nonasbestiform Analogs
| Asbestiform | Nonasbestiform |
|---|---|
| SERPENTINE | |
| Chrysotile | Antigorite, lizardite |
| AMPHIBOLE | |
| Anthophyllite asbestos | Anthophyllite |
| Cummingtonite-grunerite asbestos (“Amosite”) | Cummingtonite-grunerite |
| Crocidolite | Riebeckite |
| Tremolite asbestos | Tremolite |
| Actinolite asbestos | Actinolite |
Table 2-2 - Principal Lattice Spacings of Asbestiform Minerals a
| Minerals | Principal d-spacings (Å) and relative intensities | JCPDS Powder diffraction file 3 number | ||
|---|---|---|---|---|
| Chrysotile | 7.37100 7.36100 7.10100 |
3.6570 3.6680 2.3380 |
4.5750 2.4565 3.5570 |
21-543 b 25-645 22-1162 (theoretical) |
| “Amosite” | 8.33100 8.22100 |
3.0670 3.06085 |
2.75670 3.2570 |
17-745 (nonfibrous) 27-1170 (UICC) |
| Anthophyllite | 3.05100 3.06100 |
3.2460 8.3370 |
8.2655 3.2350 |
9-455 16-401 (synthetic) |
| Anthophyllite | 2.72100 | 2.54100 | 3.48080 | 25-157 |
| Crocidolite | 8.35100 | 3.1055 | 2.72035 | 27-1415 (UICC) |
| Tremolite | 8.38100 2.706100 3.13100 |
3.12100 3.1495 2.70660 |
2.70590 8.4340 8.4440 |
13-437 b 20-1310 b (synthetic) 23-666 (synthetic mixture with richterite) |
a This information is intended as a guide, only. Complete powder diffraction data, including mineral type and source, should be referred to, to ensure comparability of sample and reference materials where possible. Additional precision XRD data on amosite, crocidolite, tremolite, and chrysotile are available from the U.S. Bureaus of Mines. 4
b Fibrosity questionable.
Accurate quantitative analysis of asbestos in bulk samples by XRD is critically dependent on particle size distribution, crystallite size, preferred orientation and matrix absorption effects, and comparability of standard reference and sample materials. The most intense diffraction peak that has been shown to be free from interference by prior qualitative XRD analysis is selected for quantitation of each asbestiform mineral. A “thin-layer” method of analysis 5 6 is recommended in which, subsequent to comminution of the bulk material to ∼10 µm by suitable cryogenic milling techniques, an accurately known amount of the sample is deposited on a silver membrane filter. The mass of asbestiform material is determined by measuring the integrated area of the selected diffraction peak using a step-scanning mode, correcting for matrix absorption effects, and comparing with suitable calibration standards. Alternative “thick-layer” or bulk methods, 7 8 may be used for semiquantitative analysis.
This XRD method is applicable as a confirmatory method for identification and quantitation of asbestos in bulk material samples that have undergone prior analysis by PLM or other optical methods.
2.2 Range and SensitivityThe range of the method has not been determined.
The sensitivity of the method has not been determined. It will be variable and dependent upon many factors, including matrix effects (absoprtion and interferences), diagnostic reflections selected, and their relative intensities.
2.3 Limitations 2.3.1 InterferencesSince the fibrous and nonfibrous forms of the serpentine and amphibole minerals (Table 2-1) are indistinguishable by XRD techniques unless special sample preparation techniques and instrumentation are used, 9 the presence of nonasbestiform serpentines and amphiboles in a sample will pose severe interference problems in the identification and quantitative analysis of their asbestiform analogs.
The use of XRD for identification and quantitation of asbestiform minerals in bulk samples may also be limited by the presence of other interfering materials in the sample. For naturally occurring materials the commonly associated asbestos-related mineral interferences can usually be anticipated. However, for fabricated materials the nature of the interferences may vary greatly (Table 2-3) and present more serious problems in identification and quantitation. 10 Potential interferences are summarized in Table 2-4 and include the following:
• Chlorite has major peaks at 7.19 Å and 3.58 Å That interfere with both the primary (7.36 Å) and secondary (3.66 Å) peaks for chrysotile. Resolution of the primary peak to give good quantitative results may be possible when a step-scanning mode of operation is employed. • Halloysite has a peak at 3.63 Å that interferes with the secondary (3.66 Å) peak for chrysotile. • Kaolinite has a major peak at 7.15 Å that may interfere with the primary peak of chrysotile at 7.36 Å when present at concentrations of >10 percent. However, the secondary chrysotile peak at 3.66 Å may be used for quantitation. • Gypsum has a major peak at 7.5 Å that overlaps the 7.36 Å peak of chrysotile when present as a major sample constituent. This may be removed by careful washing with distilled water, or be heating to 300 °C to convert gypsum to plaster of paris. • Cellulose has a broad peak that partially overlaps the secondary (3.66 Å) chrysotile peak. 8 • Overlap of major diagnostic peaks of the amphibole asbestos minerals, amosite, anthophyllite, crocidolite, and tremolite, at approximately 8.3 Å and 3.1 Å causes mutual interference when these minerals occur in the presence of one another. In some instances, adequate resolution may be attained by using step-scanning methods and/or by decreasing the collimator slit width at the X-ray port. Table 2-3 - Common Constituents in Insulation and Wall Materials A. Insulation materialsChrysotile
“Amosite”
Crocidolite
*Rock wool
*Slag wool
*Fiber glass
Gypsum (CaSO4 · 2H2O)
Vermiculite (micas)
*Perlite
Clays (kaolin)
*Wood pulp
*Paper fibers (talc, clay, carbonate fillers)
Calcium silicates (synthetic)
Opaques (chromite, magnetite inclusions in serpentine)
Hematite (inclusions in “amosite”)
Magnesite
*Diatomaceous earth
B. Spray finishes or paintsBassanite
Carbonate minerals (calcite, dolomite, vaterite)
Talc
Tremolite
Anthophyllite
Serpentine (including chrysotile)
Amosite
Crocidolite
*Mineral wool
*Rock wool
*Slag wool
*Fiber glass
Clays (kaolin)
Micas
Chlorite
Gypsum (CaSO4 · 2H2O)
Quartz
*Organic binders and thickeners
Hyrdomagnesite
Wollastonite
Opaques (chromite, magnetite inclusions in serpentine)
Hematite (inclusions in “amosite”)
*Amorphous materials__contribute only to overall scattered radiation and increased background radiation.
Table 2-4 - Interferences in XRD Analysis Asbestiform Minerals
| Asbestiform mineral | Primary diagnostic peaks (approximate d-spacings, in Å) | Interference |
|---|---|---|
| Serpentine | ||
| Chrysotile | 7.4 | Nonasbestiform serpentines
(antigorite, lizardite) Chlorite Kaolinite Gypsum |
| 3.7 | Chlorite Halloysite Cellulose |
|
| Amphibole | ||
| “Amosite” Anthophyllite } Crocidolite Tremolite |
3.1 | Nonasbestiform amphiboles
(cummingtonite-grunerite, anthophyllite, riebeckite, tremolite) Mutual interferences Carbonates Talc |
| 8.3 | Mutual interferences |
The problem of intraspecies and matrix interferences is further aggravated by the variability of the silicate mineral powder diffraction patterns themselves, which often makes definitive identification of the asbestos minerals by comparison with standard reference diffraction patterns difficult. This variability results from alterations in the crystal lattice associated with differences in isomorphous substitution and degree of crystallinity. This is especially true for the amphiboles. These minerals exhibit a wide variety of very similar chemical compositions, with the result being that their diffraction patterns are chracterized by having major (110) reflections of the monoclinic amphiboles and (210) reflections of the orthorhombic anthophyllite separated by less than 0.2 Å. 12
2.3.2 Matrix EffectsIf a copper X-ray source is used, the presence of iron at high concentrations in a sample will result in significant X-ray fluorescence, leading to loss of peak intensity along with increased background intensity and an overall decrease in sensitivity. This situation may be corrected by choosing an X-ray source other than copper; however, this is often accompanied both by loss of intensity and by decreased resolution of closely spaced reflections. Alternatively, use of a diffracted beam monochromator will reduce background fluorescent raditation, enabling weaker diffraction peaks to be detected.
X-ray absorption by the sample matrix will result in overall attenuation of the diffracted beam and may seriously interfere with quantitative analysis. Absorption effects may be minimized by using sufficiently “thin” samples for analysis. 5 13 14 However, unless absorption effects are known to be the same for both samples and standards, appropriate corrections should be made by referencing diagnostic peak areas to an internal standard 7 8 or filter substrate (Ag) peak. 5 6
2.3.3 Particle Size DependenceBecause the intensity of diffracted X-radiation is particle-size dependent, it is essential for accurate quantitative analysis that both sample and standard reference materials have similar particle size distributions. The optimum particle size range for quantitative analysis of asbestos by XRD has been reported to be 1 to 10 µm. 15 Comparability of sample and standard reference material particle size distributions should be verified by optical microscopy (or another suitable method) prior to analysis.
2.3.4 Preferred Orientation EffectsPreferred orientation of asbestiform minerals during sample preparation often poses a serious problem in quantitative analysis by XRD. A number of techniques have been developed for reducing preferred orientation effects in “thick layer” samples. 7 8 15 However, for “thin” samples on membrane filters, the preferred orientation effects seem to be both reproducible and favorable to enhancement of the principal diagnostic reflections of asbestos minerals, actually increasing the overall sensitivity of the method. 12 14 (Further investigation into preferred orientation effects in both thin layer and bulk samples is required.)
2.3.5 Lack of Suitably Characterized Standard MaterialsThe problem of obtaining and characterizing suitable reference materials for asbestos analysis is clearly recognized. NIOSH has recently directed a major research effort toward the preparation and characterization of analytical reference materials, including asbestos standards; 16 17 however, these are not available in large quantities for routine analysis.
In addition, the problem of ensuring the comparability of standard reference and sample materials, particularly regarding crystallite size, particle size distribution, and degree of crystallinity, has yet to be adequately addressed. For example, Langer et al. 18 have observed that in insulating matrices, chrysotile tends to break open into bundles more frequently than amphiboles. This results in a line-broadening effect with a resultant decrease in sensitivity. Unless this effect is the same for both standard and sample materials, the amount of chrysotile in the sample will be underestimated by XRD analysis. To minimize this problem, it is recommended that standardized matrix reduction procedures be used for both sample and standard materials.
2.4 Precision and AccuracyPrecision of the method has not been determined.
Accuracy of the method has not been determined.
2.5 Apparatus 2.5.1 Sample PreparationSample preparation apparatus requirements will depend upon the sample type under consideration and the kind of XRD analysis to be performed.
• Mortar and Pestle: Agate or porcelain. • Razor Blades • Sample Mill: SPEX, Inc., freezer mill or equivalent. • Bulk Sample Holders • Silver Membrane Filters: 25-mm diameter, 0.45-µm pore size. Selas Corp. of America, Flotronics Div., 1957 Pioneer Road, Huntington Valley, PA 19006. • Microscope Slides • Vacuum Filtration Apparatus: Gelman No. 1107 or equivalent, and side-arm vacuum flask. • Microbalance • Ultrasonic Bath or Probe: Model W140, Ultrasonics, Inc., operated at a power density of approximately 0.1 W/mL, or equivalent. • Volumetric Flasks: 1-L volume. • Assorted Pipettes • Pipette Bulb • Nonserrated Forceps • Polyethylene Wash Bottle • Pyrex Beakers: 50-mL volume. • Desiccator • Filter Storage Cassettes • Magnetic Stirring Plate and Bars • Porcelain Crucibles • Muffle Furnace or Low Temperature Asher 2.5.2 Sample AnalysisSample analysis requirements include an X-ray diffraction unit, equipped with:
• Constant Potential Generator; Voltage and mA Stabilizers • Automated Diffractometer with Step-Scanning Mode • Copper Target X-Ray Tube: High intensity, fine focus, preferably. • X-Ray Pulse Height Selector • X-Ray Detector (with high voltage power supply): Scintillation or proportional counter. • Focusing Graphite Crystal Monochromator; or Nickel Filter (if copper source is used, and iron fluorescence is not a serious problem). • Data Output Accessories: • Strip Chart Recorder • Decade Scaler/Timer • Digital Printer • Sample Spinner (optional). • Instrument Calibration Reference Specimen: α-quartz reference crystal (Arkansas quartz standard, #180-147-00, Philips Electronics Instruments, Inc., 85 McKee Drive, Mahwah, NJ 07430) or equivalent. 2.6 Reagents 2.6.1 Standard Reference MaterialsThe reference materials listed below are intended to serve as a guide. Every attempt should be made to acquire pure reference materials that are comparable to sample materials being analyzed.
• Chrysotile: UICC Canadian, or NIEHS Plastibest. (UICC reference materials available from: UICC, MRC Pneumoconiosis Unit, Llandough Hospital, Penarth, Glamorgan, CF61XW, UK). • Crocidolite: UICC • Amosite: UICC • Anthophyllite: UICC • Tremolite Asbestos: Wards Natural Science Establishment, Rochester, N.Y.; Cyprus Research Standard, Cyprus Research, 2435 Military Ave., Los Angeles, CA 90064 (washed with dilute HCl to remove small amount of calcite impurity); India tremolite, Rajasthan State, India. • Actinolite Asbestos 2.6.2 AdhesiveTape, petroleum jelly, etc. (for attaching silver membrane filters to sample holders).
2.6.3 Surfactant1 percent aerosol OT aqueous solution or equivalent.
2.6.4 IsopropanolACS Reagent Grade.
2.7 Procedure 2.7.1 SamplingSamples for analysis of asbestos content shall be collected as specified in EPA Guidance Document #C0090, Asbestos-Containing Materials in School Buildings. 10
2.7.2 AnalysisAll samples must be analyzed initially for asbestos content by PLM. XRD should be used as an auxiliary method when a second, independent analysis is requested.
Note:Asbestos is a toxic substance. All handling of dry materials should be performed in an operating fume hood.
2.7.2.1 Sample PreparationThe method of sample preparation required for XRD analysis will depend on: (1) The condition of the sample received (sample size, homogeneity, particle size distribution, and overall composition as determined by PLM); and (2) the type of XRD analysis to be performed (qualitative, quantitative, thin layer or bulk).
Bulk materials are usually received as inhomogeneous mixtures of complex composition with very wide particle size distributions. Preparation of a homogeneous, representative sample from asbestos-containing materials is particularly difficult because the fibrous nature of the asbestos minerals inhibits mechanical mixing and stirring, and because milling procedures may cause adverse lattice alterations.
A discussion of specific matrix reduction procedures is given below. Complete methods of sample preparation are detailed in Sections 2.7.2.2 and 2.7.2.3.
Note:All samples should be examined microscopically before and after each matrix reduction step to monitor changes in sample particle size, composition, and crystallinity, and to ensure sample representativeness and homogeneity for analysis.
2.7.2.1.1 Milling - Mechanical milling of asbestos materials has been shown to decrease fiber crystallinity, with a resultant decrease in diffraction intensity of the specimen; the degree of lattice alteration is related to the duration and type of milling process. 19 22 Therefore, all milling times should be kept to a minimum.
For qualitative analysis, particle size is not usually of critical importance and initial characterization of the material with a minimum of matrix reduction is often desirable to document the composition of the sample as received. Bulk samples of very large particle size (>2-3 mm) should be comminuted to ∼100 µm. A mortar and pestle can sometimes be used in size reduction of soft or loosely bound materials though this may cause matting of some samples. Such samples may be reduced by cutting with a razor blade in a mortar, or by grinding in a suitable mill (e.g., a microhammer mill or equivalent). When using a mortar for grinding or cutting, the sample should be moistened with ethanol, or some other suitable wetting agent, to minimize exposures.
For accurate, reproducible quantitative analysis, the particle size of both sample and standard materials should be reduced to ∼10 µm (see Section 2.3.3). Dry ball milling at liquid nitrogen temperatures (e.g., Spex Freezer Mill, or equivalent) for a maximum time of 10 min. is recommended to obtain satisfactory particle size distributions while protecting the integrity of the crystal lattice. 5 Bulk samples of very large particle size may require grinding in two stages for full matrix reduction to <10 µm. 8 16
Final particle size distributions should always be verified by optical microscopy or another suitable method.
2.7.2.1.2 Low temperature ashing - For materials shown by PLM to contain large amounts of gypsum, cellulosic, or other organic materials, it may be desirable to ash the samples prior to analysis to reduce background radiation or matrix interference. Since chrysotile undergoes dehydroxylation at temperatures between 550 °C and 650 °C, with subsequent transformation to forsterite, 23 24 ashing temperatures should be kept below 500 °C. Use of a low temperature asher is recommended. In all cases, calibration of the oven is essential to ensure that a maximum ashing temperature of 500 °C is not exceeded.
2.7.2.1.3 Acid leaching - Because of the interference caused by gypsum and some carbonates in the detection of asbestiform minerals by XRD (see Section 2.3.1), it may be necessary to remove these interferents by a simple acid leaching procedure prior to analysis (see Section 1.7.2.2).
2.7.2.2 Qualitative Analysis2.7.2.2.1 Initial screening of bulk material - Qualitative analysis should be performed on a representative, homogeneous portion of the sample with a minimum of sample treatment.
1. Grind and mix the sample with a mortar and pestle (or equivalent method, see Section 2.7.2.1.1.) to a final particle size sufficiently small (∼100 µm) to allow adequate packing into the sample holder.
2. Pack the sample into a standard bulk sample holder. Care should be taken to ensure that a representative portion of the milled sample is selected for analysis. Particular care should be taken to avoid possible size segregation of the sample. (Note: Use of a back-packing method 25 of bulk sample preparation may reduce preferred orientation effects.)
3. Mount the sample on the diffractometer and scan over the diagnostic peak regions for the serpentine (∼67.4 Å) and amphibole (8.2-8.5 Å) minerals (see Table 2-2). The X-ray diffraction equipment should be optimized for intensity. A slow scanning speed of 1° 2θ/min is recommended for adequate resolution. Use of a sample spinner is recommended.
4. Submit all samples that exhibit diffraction peaks in the diagnostic regions for asbestiform minerals to a full qualitative XRD scan (5°-60° 2θ; 1°2θ/min) to verify initial peak assignments and to identify potential matrix interferences when subsequent quantitative analysis is to be performed.
5. Compare the sample XRD pattern with standard reference powder diffraction patterns (i.e., JCPDS powder diffraction data 3 or those of other well-characterized reference materials). Principal lattice spacings of asbestiform minerals are given in Table 2-2; common constituents of bulk insulation and wall materials are listed in Table 2-3.
2.7.2.2.2 Detection of minor or trace constituents - Routine screening of bulk materials by XRD may fail to detect small concentrations (<5 percent) of asbestos. The limits of detection will, in general, be improved if matrix absorption effects are minimized, and if the sample particle size is reduced to the optimal 1 to 10 µm range, provided that the crystal lattice is not degraded in the milling process. Therefore, in those instances where confirmation of the presence of an asbestiform mineral at very low levels is required, or where a negative result from initial screening of the bulk material by XRD (see Section 2.7.2.2.1) is in conflict with previous PLM results, it may be desirable to prepare the sample as described for quantitative analysis (see Section 2.7.2.3) and step-scan over appropriate 2θ ranges of selected diagnostic peaks (Table 2-2). Accurate transfer of the sample to the silver membrane filter is not necessary unless subsequent quantitative analysis is to be performed.
2.7.2.3 Quantitative AnalysisThe proposed method for quantitation of asbestos in bulk samples is a modification of the NIOSH-recommended thin-layer method for chrysotile in air. 5 A thick-layer or bulk method involving pelletizing the sample may be used for semiquantitative analysis; 7 8 however, this method requires the addition of an internal standard, use of a specially fabricated sample press, and relatively large amounts of standard reference materials. Additional research is required to evaluate the comparability of thin- and thick-layer methods for quantitative asbestos analysis.
For quantitative analysis by thin-layer methods, the following procedure is recommended:
1. Mill and size all or a substantial representative portion of the sample as outlined in Section 2.7.2.1.1.
2. Dry at 100 °C for 2 hr; cool in a desiccator.
3. Weigh accurately to the nearest 0.01 mg.
4. Samples shown by PLM to contain large amounts of cellulosic or other organic materials, gypsum, or carbonates, should be submitted to appropriate matrix reduction procedures described in Sections 2.7.2.1.2 and 2.7.2.1.3. After ashing and/or acid treatment, repeat the drying and weighing procedures described above, and determine the percent weight loss; L.
5. Quantitatively transfer an accurately weighed amount (50-100 mg) of the sample to a 1-L volumetric flask with approximately 200 mL isopropanol to which 3 to 4 drops of surfactant have been added.
6. Ultrasonicate for 10 min at a power density of approximately 0.1 W/mL, to disperse the sample material.
7. Dilute to volume with isopropanol.
8. Place flask on a magnetic stirring plate. Stir.
9. Place a silver membrane filter on the filtration apparatus, apply a vacuum, and attach the reservoir. Release the vacuum and add several milliliters of isopropanol to the reservoir. Vigorously hand shake the asbestos suspension and immediately withdraw an aliquot from the center of the suspension so that total sample weight, WT, on the filter will be approximately 1 mg. Do not adjust the volume in the pipet by expelling part of the suspension; if more than the desired aliquot is withdrawn, discard the aliquot and resume the procedure with a clean pipet. Transfer the aliquot to the reservoir. Filter rapidly under vacuum. Do not wash the reservoir walls. Leave the filter apparatus under vacuum until dry. Remove the reservoir, release the vacuum, and remove the filter with forceps. (Note: Water-soluble matrix interferences such as gypsum may be removed at this time by careful washing of the filtrate with distilled water. Extreme care should be taken not to disturb the sample.)
10. Attach the filter to a flat holder with a suitable adhesive and place on the diffractometer. Use of a sample spinner is recommended.
11. For each asbestos mineral to be quantitated select a reflection (or reflections) that has been shown to be free from interferences by prior PLM or qualitative XRD analysis and that can be used unambiguously as an index of the amount of material present in the sample (see Table 2-2).
12. Analyze the selected diagnostic reflection(s) by step scanning in increments of 0.02° 2θ for an appropriate fixed time and integrating the counts. (A fixed count scan may be used alternatively; however, the method chosen should be used consistently for all samples and standards.) An appropriate scanning interval should be selected for each peak, and background corrections made. For a fixed time scan, measure the background on each side of the peak for one-half the peak-scanning time. The net intensity, Ia, is the difference between the peak integrated count and the total background count.
13. Determine the net count, IAg, of the filter 2.36 Å silver peak following the procedure in step 12. Remove the filter from the holder, reverse it, and reattach it to the holder. Determine the net count for the unattenuated silver peak, IÅg. Scan times may be less for measurement of silver peaks than for sample peaks; however, they should be constant throughout the analysis.
14. Normalize all raw, net intensities (to correct for instrument instabilities) by referencing them to an external standard (e.g., the 3.34 Å peak of an α-quartz reference crystal). After each unknown is scanned, determine the net count, Ir , of the reference specimen following the procedure in step 12. Determine the normalized intensities by dividing the peak intensities by Ir :
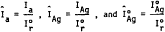 2.8
Calibration 2.8.1 Preparation of Calibration Standards
2.8
Calibration 2.8.1 Preparation of Calibration Standards
1. Mill and size standard asbestos materials according to the procedure outlined in Section 2.7.2.1.1. Equivalent, standardized matrix reduction and sizing techniques should be used for both standard and sample materials.
2. Dry at 100 °C for 2 hr; cool in a desiccator.
3. Prepare two suspensions of each standard in isopropanol by weighing approximately 10 and 50 mg of the dry material to the nearest 0.01 mg. Quantitatively transfer each to a 1-L volumetric flask with approximately 200 mL isopropanol to which a few drops of surfactant have been added.
4. Ultrasonicate for 10 min at a power density of approximately 0.1 W/mL, to disperse the asbestos material.
5. Dilute to volume with isopropanol.
6. Place the flask on a magnetic stirring plate. Stir.
7. Prepare, in triplicate, a series of at least five standard filters to cover the desired analytical range, using appropriate aliquots of the 10 and 50 mg/L suspensions and the following procedure.
Mount a silver membrane filter on the filtration apparatus. Place a few milliliters of isopropanol in the reservoir. Vigorously hand shake the asbestos suspension and immediately withdraw an aliquot from the center of the suspension. Do not adjust the volume in the pipet by expelling part of the suspension; if more than the desired aliquot is withdrawn, discard the aliquot and resume the procedure with a clean pipet. Transfer the aliquot to the reservoir. Keep the tip of the pipet near the surface of the isopropanol. Filter rapidly under vacuum. Do not wash the sides of the reservoir. Leave the vacuum on for a time sufficient to dry the filter. Release the vacuum and remove the filter with forceps.
2.8.2 Analysis of Calibration Standards1. Mount each filter on a flat holder. Perform step scans on selected diagnostic reflections of the standards and reference specimen using the procedure outlined in Section 2.7.2.3, step 12, and the same conditions as those used for the samples.
2. Determine the normalized intensity for each peak measured, Îs td, as outlined in Section 2.7.2.3, step 14.
2.9 CalculationsFor each asbestos reference material, calculate the exact weight deposited on each standard filter from the concentrations of the standard suspensions and aliquot volumes. Record the weight, w, of each standard. Prepare a calibration curve by regressing Î2s td on w. Poor reproducibility (±15 percent RSD) at any given level indicates problems in the sample preparation technique, and a need for new standards. The data should fit a straight line equation.
Determine the slope, m, of the calibration curve in counts/microgram. The intercept, b, of the line with the Îs td axis should be approximately zero. A large negative intercept indicates an error in determining the background. This may arise from incorrectly measuring the baseline or from interference by another phase at the angle of background measurement. A large positive intercept indicates an error in determining the baseline or that an impurity is included in the measured peak.
Using the normalized intensity, ÎAg, for the attenuated silver peak of a sample, and the corresponding normalized intensity from the unattenuated silver peak, ÎÅg, of the sample filter, calculate the transmittance, T, for each sample as follows: 26 27
Determine the correction factor, f(T), for each sample according to the formula:
| -R (ln T) | |||
| f (T) =__ | |||
| l-T R |
| sin ΘAg | |||
| R = | ____ | ||
| sin Θa |
Calculate the weight, Wa, in micrograms, of the asbestos material analyzed for in each sample, using the appropriate calibration data and absorption corrections:
Calculate the percent composition, Pa, of each asbestos mineral analyzed for in the parent material, from the total sample weight, WT, on the filter:
| Wa(1-.01L) | |||
| Pa = | ____ - | x 100 | |
| WT |
1. H. P. Klug and L. E. Alexander, X-ray Diffraction Procedures for Polycrystalline and Amorphous Materials, 2nd ed., New York: John Wiley and Sons, 1979.
2. L. V. Azaroff and M. J. Buerger, The Powder Method of X-ray Crystallography, New York: McGraw-Hill, 1958.
3. JCPDS-International Center for Diffraction Data Powder Diffraction File, U.S. Department of Commerce, National Bureau of Standards, and Joint Committee on Powder Diffraction Studies, Swarthmore, PA.
4. W. J. Campbell, C. W. Huggins, and A. G. Wylie, Chemical and Physical Characterization of Amosite, Chrysotile, Crocidolite, and Nonfibrous Tremolite for National Institute of Environmental Health Sciences Oral Ingestion Studies, U.S. Bureau of Mines Report of Investigation RI8452, 1980.
5. B. A. Lange and J. C. Haartz, Determination of microgram quantities of asbestos by X-ray diffraction: Chrysotile in thin dust layers of matrix material, Anal. Chem., 51(4):520-525, 1979.
6. NIOSH Manual of Analytical Methods, Volume 5, U.S. Dept. HEW, August 1979, pp. 309-1 to 309-9.
7. H. Dunn and J. H. Stewart, Jr., Quantitative determination of chrysotile in building materials, The Microscope, 29(1), 1981.
8. M. Taylor, Methods for the quantitative determination of asbestos and quartz in bulk samples using X-ray diffraction, The Analyst, 103(1231):1009-1020, 1978.
9. L. Birks, M. Fatemi, J. V. Gilfrich, and E. T. Johnson, Quantitative Analysis of Airborne Asbestos by X-ray Diffraction, Naval Research Laboratory Report 7879, Naval Research Laboratory, Washington, DC, 1975.
10. U.S. Environmental Protection Agency, Asbestos-Containing Materials in School Buildings: A Guidance Document, Parts 1 and 2, EPA/OPPT No. C00090, March 1979.
11. J. B. Krause and W. H. Ashton, Misidentification of asbestos in talc, pp. 339-353, in: Proceedings of Workshop on Asbestos: Definitions and Measurement Methods (NBS Special Publication 506), C. C. Gravatt, P. D. LaFleur, and K. F. Heinrich (eds.), Washington, DC: National Measurement Laboratory, National Bureau of Standards, 1977 (issued 1978).
12. H. D. Stanley, The detection and identification of asbestos and asbesti-form minerals in talc, pp. 325-337, in Proceedings of Workshop on Asbestos: Definitions and Measurement Methods (NBS Special Publication 506), C. C. Gravatt, P. D. LaFleur, and K. F. Heinrich (eds.), Washington, DC, National Measurement Laboratory, National Bureau of Standards, 1977 (issued 1978).
13. A. L. Rickards, Estimation of trace amounts of chrysotile asbestos by X-ray diffraction, Anal. Chem., 44(11):1872-3, 1972.
14. P. M. Cook, P. L. Smith, and D. G. Wilson, Amphibole fiber concentration and determination for a series of community air samples: use of X-ray diffraction to supplement electron microscope analysis, in: Electron Microscopy and X-ray Applications to Environmental and Occupation Health Analysis, P. A. Russell and A. E. Hutchings (eds.), Ann Arbor: Ann Arbor Science Publications, 1977.
15. A. N. Rohl and A. M. Langer, Identification and quantitation of asbestos in talc, Environ. Health Perspectives, 9:95-109, 1974.
16. J. L. Graf, P. K. Ase, and R. G. Draftz, Preparation and Characterization of Analytical Reference Minerals, DHEW (NIOSH) Publication No. 79-139, June 1979.
17. J. C. Haartz, B. A. Lange, R. G. Draftz, and R. F. Scholl, Selection and characterization of fibrous and nonfibrous amphiboles for analytical methods development, pp. 295-312, in: Proceedings of Workshop on Asbestos: Definitions and Measurement Methods (NBS Special Publication 506), C. C. Gravatt, P. D. LaFleur, and K. F. Heinrich (eds.), Washington, DC: National Measurement Laboratory, National Bureau of Standards, 1977 (issued 1978).
18. Personal communication, A. M. Langer, Environmental Sciences Laboratory, Mount Sinai School of Medicine of the City University of New York, New York, New York.
19. A. M. Langer, M. S. Wolff, A. N. Rohl, and I. J. Selikoff, Variation of properties of chrysotile asbestos subjected to milling, J. Toxicol. and Environ. Health, 4:173-188, 1978.
20. A. M. Langer, A. D. Mackler, and F. D. Pooley, Electron microscopical investigation of asbestos fibers, Environ. Health Perspect., 9:63-80, 1974.
21. E. Occella and G. Maddalon, X-ray diffraction characteristics of some types of asbestos in relation to different techniques of comminution, Med. Lavoro, 54(10):628-636, 1963.
22. K. R. Spurny, W. Stöber, H. Opiela, and G. Weiss, On the problem of milling and ultrasonic treatment of asbestos and glass fibers in biological and analytical applications, Am. Ind. Hyg. Assoc. J., 41:198-203, 1980.
23. L. G. Berry and B. Mason, Mineralogy, San Francisco: W. H. Greeman & Co., 1959.
24. J. P. Schelz, The detection of chrysotile asbestos at low levels in talc by differential thermal analysis, Thermochimica Acta, 8:197-204, 1974.
25. Reference 1, pp. 372-374.
26. J. Leroux, Staub-Reinhalt Luft, 29:26 (English), 1969.
27. J. A. Leroux, B. C. Davey, and A. Paillard, Am. Ind. Hyg. Assoc. J., 34:409, 1973.
[47 FR 23369, May 27, 1982; 47 FR 38535, Sept. 1, 1982; Redesignated at 60 FR 31922, June 19, 1995]