Title 40
PART 61 APPENDIX
| Vessel capacity (cubic meters) |
Vapor pressure 1 (kilopascals) |
|---|---|
| 75 ≤capacity <151 | ≥13.1 |
| 151 ≤capacity | ≥5.2 |
1 Maximum true vapor pressure as defined in § 61.241.
Table 2 to Subpart V of Part 61 - Surge Control Vessels and Bottoms Receivers at New Sources
40:10.0.1.1.1.22.1.20.3 :
Table 2 to Subpart V of Part 61 - Surge Control Vessels and Bottoms Receivers at New Sources| Vessel capacity (cubic meters) |
Vapor pressure 1 (kilopascals) |
|---|---|
| 38 ≤capacity <151 | ≥13.1 |
| 151 ≤capacity | ≥0.7 |
1 Maximum true vapor pressure as defined in § 61.241.
Appendix A to Part 61
40:10.0.1.1.1.29.1.21.4 : Appendix A
Appendix A to Part 61





 II. Waiver
Requests
II. Waiver
Requests
A. Waiver of Compliance. Owners or operators of sources unable to operate in compliance with the National Emission Standards for Hazardous Air Pollutants prior to 90 days after the effective date of any standards or amendments which require the submission of such information may request a waiver of compliance from the Administrator of the U.S. Environmental Protection Agency for the time period necessary to install appropriate control devices or make modifications to achieve compliance. The Administrator may grant a waiver of compliance with the standard for a period not exceeding two years from the effective date of the hazardous pollutant standards, if he finds that such period is necessary for the installation of controls and that steps will be taken during the period of the waiver to assure that the health of persons will be protected from imminent endangerment.
The report information provided in Section I must accompany this application. Applications should be sent to the appropriate EPA regional office.1. Processes Involved - Indicate the process or processes emitting hazardous pollutants to which emission controls are to be applied.
2. Controls
a. Describe the proposed type of control device to be added or modification to be made to the process to reduce the emission of hazardous pollutants to an acceptable level. (Use additional sheets if necessary.)
b. Describe the measures that will be taken during the waiver period to assure that the health of persons will be protected from imminent endangerment. (Use additional sheets if necessary.)
3. Increments of Progress - Specify the dates by which the following increments of progress will be met.
Date by which contracts for emission control systems or process modifications will be awarded; or date by which orders will be issued for the purchase of the component parts to accomplish emission control or process modification.
B. Waiver of Emission Tests. A waiver of emission testing may be granted to owners or operators of sources subject to emission testing if, in the judgment of the Administrator of the Environmental Protection Agency the emissions from the source comply with the appropriate standard or if the owners or operators of the source have requested a waiver of compliance or have been granted a waiver of compliance.
This application should accompany the report information provided in Section I.1. Reason - State the reasons for requesting a waiver of emission testing. If the reason stated is that the emissions from the source are within the prescribed limits, documentation of this condition must be attached.
Date Signature of the owner or operator (Sec. 114, of the Clean Air Act as amended (42 U.S.C. 7414)) [40 FR 48303, Oct. 14, 1975, as amended at 43 FR 8800, Mar. 3, 1978; 50 FR 46295, Sept. 9, 1985]Appendix B to Part 61 - Test Methods
40:10.0.1.1.1.29.1.21.5 : Appendix B
Appendix B to Part 61 - Test Methods Method 101 - Determination of particulate and gaseous mercury emissions from chlor-alkali plants (air streams) Method 101A - Determination of particulate and gaseous mercury emissions from sewage sludge incinerators Method 102 - Determination of particulate and gaseous mercury emissions from chlor-alkali plants (hydrogen streams) Method 103 - Beryllium screening method Method 104 - Determination of beryllium emissions from stationary sources Method 105 - Determination of mercury in wastewater treatment plant sewage sludges Method 106 - Determination of vinyl chloride emissions from stationary sources Method 107 - Determination of vinyl chloride content of in-process wastewater samples, and vinyl chloride content of polyvinyl chloride resin slurry, wet cake, and latex samples Method 107A - Determination of vinyl chloride content of solvents, resin-solvent solution, polyvinyl chloride resin, resin slurry, wet resin, and latex samples Method 108 - Determination of particulate and gaseous arsenic emissions Method 108A - Determination of arsenic content in ore samples from nonferrous smelters Method 108B - Determination of arsenic content in ore samples from nonferrous smelters Method 108C - Determination of arsenic content in ore samples from nonferrous smelters (molybdenum blue photometric procedure) Method 111 - Determination of Polonium - 210 emissions from stationary sources Method 114 - Test Methods for Measuring Radionuclide Emissions From Stationary Sources Method 115 - Monitoring for Radon-222 Emissions Method 101 - Determination of Particulate and Gaseous Mercury Emissions From Chlor-Alkali Plants (Air Streams) Note:This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from methods in appendix A to 40 CFR part 60. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, and Method 5.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Mercury (Hg) | 7439-97-6 | Dependent upon recorder and spectrophotometer. |
1.2 Applicability. This method is applicable for the determination of Hg emissions, including both particulate and gaseous Hg, from chlor-alkali plants and other sources (as specified in the regulations) where the carrier-gas stream in the duct or stack is principally air.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of MethodParticulate and gaseous Hg emissions are withdrawn isokinetically from the source and collected in acidic iodine monochloride (ICl) solution. The Hg collected (in the mercuric form) is reduced to elemental Hg, which is then aerated from the solution into an optical cell and measured by atomic absorption spectrophotometry.
3.0 Definitions [Reserved] 4.0 Interferences4.1 Sample Collection. Sulfur dioxide (SO2) reduces ICl and causes premature depletion of the ICl solution.
4.2 Sample Analysis.
4.2.1 ICl concentrations greater than 10−4 molar inhibit the reduction of the Hg (II) ion in the aeration cell.
4.2.2 Condensation of water vapor on the optical cell windows causes a positive interference.
5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method does not purport to address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burn as thermal burn.
5.2.1 Hydrochloric Acid (HCl). Highly toxic and corrosive. Causes severe damage to tissues. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Nitric Acid (HNO3). Highly corrosive to eyes, skin, nose, and lungs. Vapors cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.3 Sulfuric Acid (H2SO4). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. 3 mg/m 3 will cause lung damage. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0 Equipment and Supplies.6.1 Sample Collection. A schematic of the sampling train used in performing this method is shown in Figure 101-1; it is similar to the Method 5 sampling train. The following items are required for sample collection:
6.1.1 Probe Nozzle, Pitot Tube, Differential Pressure Gauge, Metering System, Barometer, and Gas Density Determination Equipment. Same as Method 5, Sections 6.1.1.1, 6.1.1.3, 6.1.1.4, 6.1.1.9, 6.1.2, and 6.1.3, respectively.
6.1.2 Probe Liner. Borosilicate or quartz glass tubing. A heating system capable of maintaining a gas temperature of 120 ±14 °C (248 ±25 °F) at the probe exit during sampling may be used to prevent water condensation.
Note:Do not use metal probe liners.
6.1.3 Impingers. Four Greenburg-Smith impingers connected in series with leak-free ground glass fittings or any similar leak-free noncontaminating fittings. For the first, third, and fourth impingers, impingers that are modified by replacing the tip with a 13-mm ID (0.5-in.) glass tube extending to 13 mm (0.5 in.) from the bottom of the flask may be used.
6.1.4 Acid Trap. Mine Safety Appliances air line filter, Catalog number 81857, with acid absorbing cartridge and suitable connections, or equivalent.
6.2 Sample Recovery. The following items are needed for sample recovery:
6.2.1 Glass Sample Bottles. Leakless, with Teflon-lined caps, 1000- and 100-ml.
6.2.2 Graduated Cylinder. 250-ml.
6.2.3 Funnel and Rubber Policeman. To aid in transfer of silica gel to container; not necessary if silica gel is weighed in the field.
6.2.4 Funnel. Glass, to aid in sample recovery.
6.3 Sample Preparation and Analysis. The following items are needed for sample preparation and analysis:
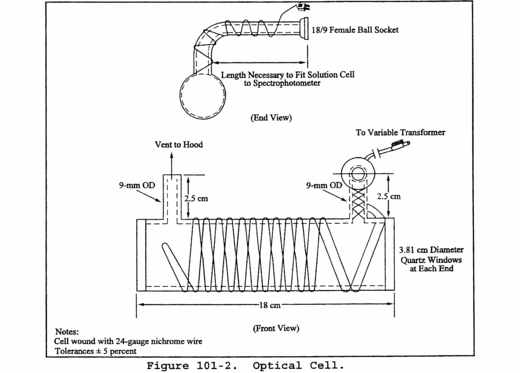
6.3.1 Atomic Absorption Spectrophotometer. Perkin-Elmer 303, or equivalent, containing a hollow-cathode mercury lamp and the optical cell described in Section 6.3.2.
6.3.2 Optical Cell. Cylindrical shape with quartz end windows and having the dimensions shown in Figure 101-2. Wind the cell with approximately 2 meters (6 ft) of 24-gauge Nichrome wire, or equivalent, and wrap with fiberglass insulation tape, or equivalent; do not let the wires touch each other.
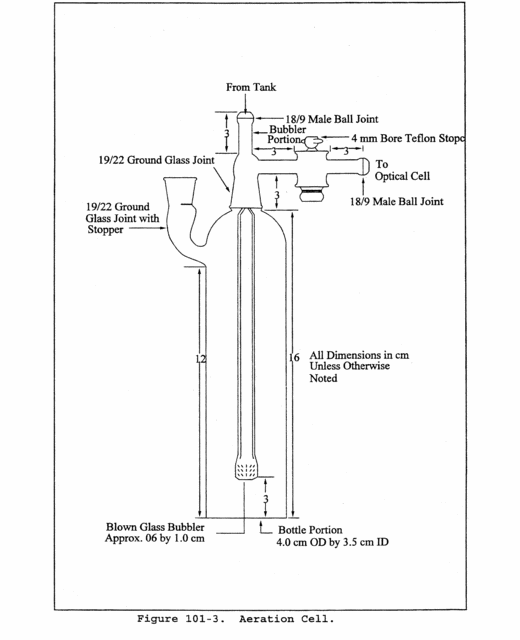
6.3.3 Aeration Cell. Constructed according to the specifications in Figure 101-3. Do not use a glass frit as a substitute for the blown glass bubbler tip shown in Figure 101-3.
6.3.4 Recorder. Matched to output of the spectrophotometer described in Section 6.3.1.
6.3.5 Variable Transformer. To vary the voltage on the optical cell from 0 to 40 volts.
6.3.6 Hood. For venting optical cell exhaust.
6.3.7 Flow Metering Valve.
6.3.8 Rate Meter. Rotameter, or equivalent, capable of measuring to within 2 percent a gas flow of 1.5 liters/min (0.053 cfm).
6.3.9 Aeration Gas Cylinder. Nitrogen or dry, Hg-free air, equipped with a single-stage regulator.
6.3.10 Tubing. For making connections. Use glass tubing (ungreased ball and socket connections are recommended) for all tubing connections between the solution cell and the optical cell; do not use Tygon tubing, other types of flexible tubing, or metal tubing as substitutes. Teflon, steel, or copper tubing may be used between the nitrogen tank and flow metering valve (Section 6.3.7), and Tygon, gum, or rubber tubing between the flow metering valve and the aeration cell.
6.3.11 Flow Rate Calibration Equipment. Bubble flow meter or wet-test meter for measuring a gas flow rate of 1.5 ±0.1 liters/min (0.053 ±0.0035 cfm).
6.3.12 Volumetric Flasks. Class A with penny head standard taper stoppers; 100-, 250-, 500-, and 1000-ml.
6.3.13 Volumetric Pipets. Class A; 1-, 2-, 3-, 4-, and 5-ml.
6.3.14 Graduated Cylinder. 50-ml.
6.3.15 Magnetic Stirrer. General-purpose laboratory type.
6.3.16 Magnetic Stirring Bar. Teflon-coated.
6.3.17 Balance. Capable of weighing to ±0.5 g.
6.3.18 Alternative Analytical Apparatus. Alternative systems are allowable as long as they meet the following criteria:
6.3.18.1 A linear calibration curve is generated and two consecutive samples of the same aliquot size and concentration agree within 3 percent of their average.
6.3.18.2 A minimum of 95 percent of the spike is recovered when an aliquot of a source sample is spiked with a known concentration of Hg (II) compound.
6.3.18.3 The reducing agent should be added after the aeration cell is closed.
6.3.18.4 The aeration bottle bubbler should not contain a frit.
6.3.18.5 Any Tygon tubing used should be as short as possible and conditioned prior to use until blanks and standards yield linear and reproducible results.
6.3.18.6 If manual stirring is done before aeration, it should be done with the aeration cell closed.
6.3.18.7 A drying tube should not be used unless it is conditioned as the Tygon tubing above.
7.0 Reagents and StandardsUnless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society; where such specifications are not available, use the best available grade.
7.1 Sample Collection. The following reagents are required for sample collection:
7.1.1 Water. Deionized distilled, to conform to ASTM D 1193-77 or 91 (incorporated by reference - see § 61.18), Type 1. If high concentrations of organic matter are not expected to be present, the analyst may eliminate the KMnO4 test for oxidizable organic matter. Use this water in all dilutions and solution preparations.
7.1.2 Nitric Acid, 50 Percent (v/v). Mix equal volumes of concentrated HNO3 and water, being careful to add the acid to the water slowly.
7.1.3 Silica Gel. Indicating type, 6- to 16-mesh. If previously used, dry at 175 °C (350 °F) for 2 hours. The tester may use new silica gel as received.
7.1.4 Potassium Iodide (KI) Solution, 25 Percent. Dissolve 250 g of KI in water, and dilute to 1 liter.
7.1.5 Iodine Monochloride Stock Solution, 1.0 M. To 800 ml of 25 percent KI solution, add 800 ml of concentrated HCl. Cool to room temperature. With vigorous stirring, slowly add 135 g of potassium iodate (KIO3), and stir until all free iodine has dissolved. A clear orange-red solution occurs when all the KIO3 has been added. Cool to room temperature, and dilute to 1800 ml with water. Keep the solution in amber glass bottles to prevent degradation.
7.1.6 Absorbing Solution, 0.1 M ICl. Dilute 100 ml of the 1.0 M ICl stock solution to 1 liter with water. Keep the solution in amber glass bottles and in darkness to prevent degradation. This reagent is stable for at least two months.
7.2 Sample Preparation and Analysis. The following reagents and standards are required for sample preparation and analysis:
7.2.1 Reagents.
7.2.1.1 Tin (II) Solution. Prepare fresh daily, and keep sealed when not being used. Completely dissolve 20 g of tin (II) chloride (or 25 g of tin (II) sulfate) crystals (Baker Analyzed reagent grade or any other brand that will give a clear solution) in 25 ml of concentrated HCl. Dilute to 250 ml with water. Do not substitute HNO3, H2SO4, or other strong acids for the HCl.
7.2.1.2 Sulfuric Acid, 5 Percent (v/v). Dilute 25 ml of concentrated H2SO4 to 500 ml with water.
7.2.2 Standards
7.2.2.1 Hg Stock Solution, 1 mg Hg/ml. Prepare and store all Hg standard solutions in borosilicate glass containers. Completely dissolve 0.1354 g of Hg (II) chloride in 75 ml of water in a 100-ml glass volumetric flask. Add 10 ml of concentrated HNO3, and adjust the volume to exactly 100 ml with water. Mix thoroughly. This solution is stable for at least one month.
7.2.2.2 Intermediate Hg Standard Solution, 10 µg Hg/ml. Prepare fresh weekly. Pipet 5.0 ml of the Hg stock solution (Section 7.2.2.1) into a 500-ml glass volumetric flask, and add 20 ml of the 5 percent H2SO4 solution. Dilute to exactly 500 ml with water. Thoroughly mix the solution.
7.2.2.3 Working Hg Standard Solution, 200 ng Hg/ml. Prepare fresh daily. Pipet 5.0 ml of the intermediate Hg standard solution (Section 7.2.2.2) into a 250-ml volumetric glass flask. Add 10 ml of the 5 percent H2SO4 and 2 ml of the 0.1 M ICl absorbing solution taken as a blank (Section 8.7.4.3), and dilute to 250 ml with water. Mix thoroughly.
8.0 Sample Collection, Preservation, Transport, and StorageBecause of the complexity of this method, testers should be trained and experienced with the test procedures to ensure reliable results. Since the amount of Hg that is collected generally is small, the method must be carefully applied to prevent contamination or loss of sample.
8.1 Pretest Preparation. Follow the general procedure outlined in Method 5, Section 8.1, except omit Sections 8.1.2 and 8.1.3.
8.2 Preliminary Determinations. Follow the general procedure outlined in Method 5, Section 8.2, with the exception of the following:
8.2.1 Select a nozzle size based on the range of velocity heads to assure that it is not necessary to change the nozzle size in order to maintain isokinetic sampling rates below 28 liters/min (1.0 cfm).
8.2.2 Perform test runs such that samples are obtained over a period or periods that accurately determine the maximum emissions that occur in a 24-hour period. In the case of cyclic operations, run sufficient tests for the accurate determination of the emissions that occur over the duration of the cycle. A minimum sample time of 2 hours is recommended. In some instances, high Hg or high SO2 concentrations make it impossible to sample for the desired minimum time. This is indicated by reddening (liberation of free iodine) in the first impinger. In these cases, the sample run may be divided into two or more subruns to ensure that the absorbing solution is not depleted.
8.3 Preparation of Sampling Train.
8.3.1 Clean all glassware (probe, impingers, and connectors) by rinsing with 50 percent HNO3, tap water, 0.1 M ICl, tap water, and finally deionized distilled water. Place 100 ml of 0.1 M ICl in each of the first three impingers. Take care to prevent the absorbing solution from contacting any greased surfaces. Place approximately 200 g of preweighed silica gel in the fourth impinger. More silica gel may be used, but care should be taken to ensure that it is not entrained and carried out from the impinger during sampling. Place the silica gel container in a clean place for later use in the sample recovery. Alternatively, determine and record the weight of the silica gel plus impinger to the nearest 0.5 g.
8.3.2 Install the selected nozzle using a Viton A O-ring when stack temperatures are less than 260 °C (500 °F). Use a fiberglass string gasket if temperatures are higher. See APTD-0576 (Reference 3 in Method 5) for details. Other connecting systems using either 316 stainless steel or Teflon ferrules may be used. Mark the probe with heat-resistant tape or by some other method to denote the proper distance into the stack or duct for each sampling point.
8.3.3 Assemble the train as shown in Figure 101-1, using (if necessary) a very light coat of silicone grease on all ground glass joints. Grease only the outer portion (see APTD-0576) to avoid the possibility of contamination by the silicone grease.
Note:An empty impinger may be inserted between the third impinger and the silica gel to remove excess moisture from the sample stream.
8.3.4 After the sampling train has been assembled, turn on and set the probe heating system, if applicable, at the desired operating temperature. Allow time for the temperatures to stabilize. Place crushed ice around the impingers.
8.4 Leak-Check Procedures. Follow the leak-check procedures outlined in Method 5, Section 8.4.
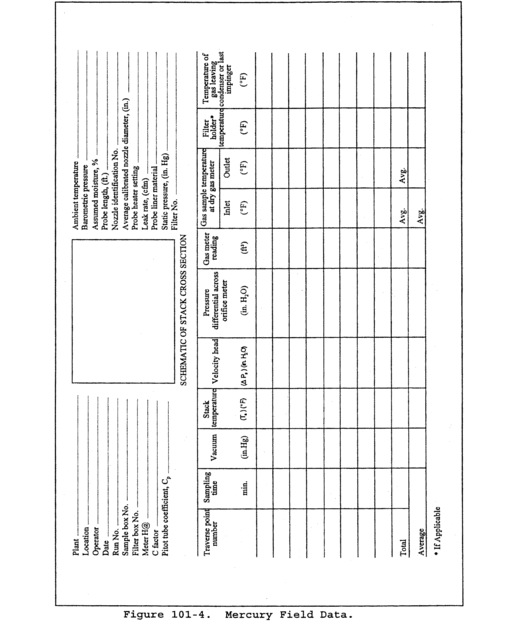
8.5 Sampling Train Operation. Follow the general procedure outlined in Method 5, Section 8.5. For each run, record the data required on a data sheet such as the one shown in Figure 101-4.
8.6 Calculation of Percent Isokinetic. Same as Method 5, Section 8.6.
8.7 Sample Recovery. Begin proper cleanup procedure as soon as the probe is removed from the stack at the end of the sampling period.
8.7.1 Allow the probe to cool. When it can be safely handled, wipe off any external particulate matter near the tip of the probe nozzle, and place a cap over it. Do not cap off the probe tip tightly while the sampling train is cooling. Capping would create a vacuum and draw liquid out from the impingers.
8.7.2 Before moving the sampling train to the cleanup site, remove the probe from the train, wipe off the silicone grease, and cap the open outlet of the probe. Be careful not to lose any condensate that might be present. Wipe off the silicone grease from the impinger. Use either ground-glass stoppers, plastic caps, or serum caps to close these openings.
8.7.3 Transfer the probe and impinger assembly to a cleanup area that is clean, protected from the wind, and free of Hg contamination. The ambient air in laboratories located in the immediate vicinity of Hg-using facilities is not normally free of Hg contamination.
8.7.4 Inspect the train before and during disassembly, and note any abnormal conditions. Treat the samples as follows.
8.7.4.1 Container No. 1 (Impingers and Probe).
8.7.4.1.1 Using a graduated cylinder, measure the liquid in the first three impingers to within 1 ml. Record the volume of liquid present (e.g., see Figure 5-6 of Method 5). This information is needed to calculate the moisture content of the effluent gas. (Use only glass storage bottles and graduated cylinders that have been precleaned as in Section 8.3.1) Place the contents of the first three impingers into a 1000-ml glass sample bottle.
8.7.4.1.2 Taking care that dust on the outside of the probe or other exterior surfaces does not get into the sample, quantitatively recover the Hg (and any condensate) from the probe nozzle, probe fitting, and probe liner as follows: Rinse these components with two 50-ml portions of 0.1 M ICl. Next, rinse the probe nozzle, fitting and liner, and each piece of connecting glassware between the probe liner and the back half of the third impinger with a maximum of 400 ml of water. Add all washings to the 1000-ml glass sample bottle containing the liquid from the first three impingers.
8.7.4.1.3 After all washings have been collected in the sample container, tighten the lid on the container to prevent leakage during shipment to the laboratory. Mark the height of the liquid to determine later whether leakage occurred during transport. Label the container to identify clearly its contents.
8.7.4.2 Container No. 2 (Silica Gel). Same as Method 5, Section 8.7.6.3.
8.7.4.3 Container No. 3 (Absorbing Solution Blank). Place 50 ml of the 0.1 M ICl absorbing solution in a 100-ml sample bottle. Seal the container. Use this blank to prepare the working Hg standard solution (Section 7.2.2.3).
9.0 Quality Control9.1 Miscellaneous Quality Control Measures.
| Section | Quality control measure | Effect |
|---|---|---|
| 8.4 10.2 | Sampling equipment leak-checks and calibration | Ensure accuracy and precision of sampling measurements. |
| 10.5, 10.6 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
| 11.3.3 | Check for matrix effects | Eliminate matrix effects. |
9.2 Volume Metering System Checks. Same as Method 5, Section 9.2.
10.0 Calibration and Standardizations Note:Maintain a laboratory log of all calibrations.
10.1 Before use, clean all glassware, both new and used, as follows: brush with soap and tap water, liberally rinse with tap water, soak for 1 hour in 50 percent HNO3, and then rinse with deionized distilled water.
10.2 Sampling Equipment. Calibrate the sampling equipment according to the procedures outlined in the following sections of Method 5: Section 10.1 (Probe Nozzle), Section 10.2 (Pitot Tube Assembly), Section 10.3 (Metering System), Section 10.5 (Temperature Sensors), Section 10.6 (Barometer).
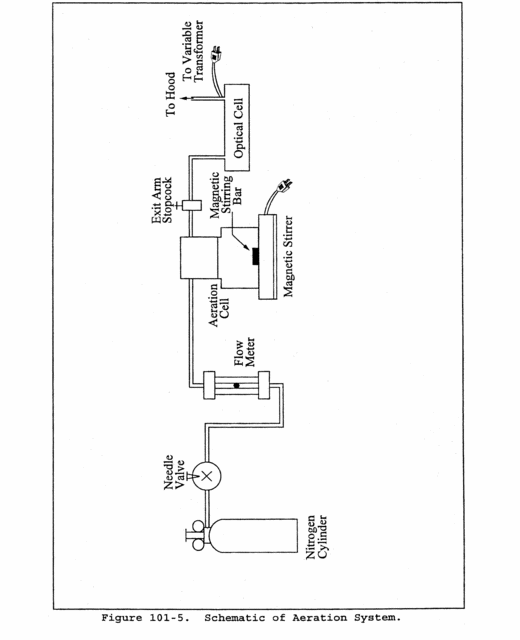
10.3 Aeration System Flow Rate Meter. Assemble the aeration system as shown in Figure 101-5. Set the outlet pressure on the aeration gas cylinder regulator to a minimum pressure of 500 mm Hg (10 psi), and use the flow metering valve and a bubble flowmeter or wet-test meter to obtain a flow rate of 1.5 ±0.1 liters/min (0.053 ±0.0035 cfm) through the aeration cell. After the calibration of the aeration system flow rate meter is complete, remove the bubble flowmeter from the system.
10.4 Optical Cell Heating System. Using a 50-ml graduated cylinder, add 50 ml of water to the bottle section of the aeration cell, and attach the bottle section to the bubbler section of the cell. Attach the aeration cell to the optical cell and while aerating at 1.5 ±0.1 liters/min (0.053 ±0.0035 cfm), determine the minimum variable transformer setting necessary to prevent condensation of moisture in the optical cell and in the connecting tubing. (This setting should not exceed 20 volts.)
10.5 Spectrophotometer and Recorder.
10.5.1 The Hg response may be measured by either peak height or peak area.
Note:The temperature of the solution affects the rate at which elemental Hg is released from a solution and, consequently, it affects the shape of the absorption curve (area) and the point of maximum absorbance (peak height). Therefore, to obtain reproducible results, bring all solutions to room temperature before use.
10.5.2 Set the spectrophotometer wavelength at 253.7 nm, and make certain the optical cell is at the minimum temperature that will prevent water condensation. Then set the recorder scale as follows: Using a 50-ml graduated cylinder, add 50 ml of water to the aeration cell bottle. Add three drops of Antifoam B to the bottle, and then pipet 5.0 ml of the working Hg standard solution into the aeration cell.
Note:Always add the Hg-containing solution to the aeration cell after the 50 ml of water.
10.5.3 Place a Teflon-coated stirring bar in the bottle. Before attaching the bottle section to the bubbler section of the aeration cell, make certain that (1) the aeration cell exit arm stopcock (Figure 101-3) is closed (so that Hg will not prematurely enter the optical cell when the reducing agent is being added) and (2) there is no flow through the bubbler. If conditions (1) and (2) are met, attach the bottle section to the bubbler section of the aeration cell. Pipet 5 ml of tin (II) reducing solution into the aeration cell through the side arm, and immediately stopper the side arm. Stir the solution for 15 seconds, turn on the recorder, open the aeration cell exit arm stopcock, and immediately initiate aeration with continued stirring. Determine the maximum absorbance of the standard, and set this value to read 90 percent of the recorder full scale.
10.6 Calibration Curve.
10.6.1 After setting the recorder scale, repeat the procedure in Section 10.5 using 0.0-, 1.0-, 2.0-, 3.0-, 4.0-, and 5.0-ml aliquots of the working standard solution (final amount of Hg in the aeration cell is 0, 200, 400, 600, 800, and 1000 ng, respectively). Repeat this procedure on each aliquot size until two consecutive peaks agree within 3 percent of their average value.
Note:To prevent Hg carryover from one sample to another, do not close the aeration cell from the optical cell until the recorder pen has returned to the baseline.)
10.6.2 It should not be necessary to disconnect the aeration gas inlet line from the aeration cell when changing samples. After separating the bottle and bubbler sections of the aeration cell, place the bubbler section into a 600-ml beaker containing approximately 400 ml of water. Rinse the bottle section of the aeration cell with a stream of water to remove all traces of the tin (II) reducing agent. Also, to prevent the loss of Hg before aeration, remove all traces of the reducing agent between samples by washing with water. It will be necessary, however, to wash the aeration cell parts with concentrated HCl if any of the following conditions occur: (1) A white film appears on any inside surface of the aeration cell, (2) the calibration curve changes suddenly, or (3) the replicate samples do not yield reproducible results.
10.6.3 Subtract the average peak height (or peak area) of the blank (0.0-ml aliquot) - which must be less than 2 percent of recorder full scale - from the averaged peak heights of the 1.0-, 2.0-, 3.0-, 4.0-, and 5.0-ml aliquot standards. If the blank absorbance is greater than 2 percent of full-scale, the probable cause is Hg contamination of a reagent or carry-over of Hg from a previous sample. Prepare the calibration curve by plotting the corrected peak height of each standard solution versus the corresponding final total Hg weight in the aeration cell (in ng), and draw the best fit straight line. This line should either pass through the origin or pass through a point no further from the origin than ±2 percent of the recorder full scale. If the line does not pass through or very near to the origin, check for nonlinearity of the curve and for incorrectly prepared standards.
11.0 Analytical Procedure11.1 Sample Loss Check. Check the liquid level in each container to see whether liquid was lost during transport. If a noticeable amount of leakage occurred, either void the sample or use methods subject to the approval of the Administrator to account for the losses.
11.2 Sample Preparation. Treat each sample as follows:
11.2.1 Container No. 1 (Impingers and Probe). Carefully transfer the contents of Container No. 1 into a 1000-ml volumetric flask, and adjust the volume to exactly 1000 ml with water.
11.2.2 Dilutions. Pipet a 2-ml aliquot from the diluted sample from Section 11.2.1 into a 250-ml volumetric flask. Add 10 ml of 5 percent H2SO4, and adjust the volume to exactly 250 ml with water. This solution is stable for at least 72 hours.
Note:The dilution factor will be 250/2 for this solution.
11.3 Analysis. Calibrate the analytical equipment and develop a calibration curve as outlined in Sections 10.3 through 10.6.
11.3.1 Mercury Samples. Repeat the procedure used to establish the calibration curve with an appropriately sized aliquot (1 to 5 ml) of the diluted sample (from Section 11.2.2) until two consecutive peak heights agree within 3 percent of their average value. The peak maximum of an aliquot (except the 5-ml aliquot) must be greater than 10 percent of the recorder full scale. If the peak maximum of a 1.0-ml aliquot is off scale on the recorder, further dilute the original source sample to bring the Hg concentration into the calibration range of the spectrophotometer.
11.3.2 Run a blank and standard at least after every five samples to check the spectrophotometer calibration. The peak height of the blank must pass through a point no further from the origin than ±2 percent of the recorder full scale. The difference between the measured concentration of the standard (the product of the corrected peak height and the reciprocal of the least squares slope) and the actual concentration of the standard must be less than 7 percent, or recalibration of the analyzer is required.
11.3.3 Check for Matrix Effects (optional). Use the Method of Standard Additions as follows to check at least one sample from each source for matrix effects on the Hg results. The Method of Standard Additions procedures described on pages 9-4 and 9-5 of the section entitled “General Information” of the Perkin Elmer Corporation Atomic Absorption Spectrophotometry Manual, Number 303-0152 (Reference 16 in Section 16.0) are recommended. If the results of the Method of Standard Additions procedure used on the single source sample do not agree to within ±5 percent of the value obtained by the routine atomic absorption analysis, then reanalyze all samples from the source using the Method of Standard Additions procedure.
11.4 Container No. 2 (Silica Gel). Weigh the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g using a balance. (This step may be conducted in the field.)
12.0 Data Analysis and CalculationsCarry out calculations, retaining at least one extra decimal significant figure beyond that of the acquired data. Round off figures only after the final calculation. Other forms of the equations may be used as long as they give equivalent results.
12.1 Average Dry Gas Meter Temperature and Average Orifice Pressure Drop, Dry Gas Volume, Volume of Water Vapor Condensed, Moisture Content, and Isokinetic Variation. Same as Method 5, Sections 12.2 through 12.5 and 12.11, respectively.
12.2 Stack Gas Velocity. Using the data from this test and Equation 2-9 of Method 2, calculate the average stack gas velocity vs.
12.3 Total Mercury.
12.3.1 For each source sample, correct the average maximum absorbance of the two consecutive samples whose peak heights agree within 3 percent of their average for the contribution of the solution blank (see Section 10.6.3). Use the calibration curve and these corrected averages to determine the final total weight of Hg in ng in the aeration cell for each source sample.
12.3.2 Correct for any dilutions made to bring the sample into the working range of the spectrophotometer. Then calculate the Hg in the original solution, mHg, as follows:
Where: CHg(AC) = Total ng of Hg in aliquot analyzed (reagent blank subtracted). DF = Dilution factor for the Hg-containing solution (before adding to the aeration cell; e.g., DF = 250/2 if the source samples were diluted as described in Section 11.2.2). Vf = Solution volume of original sample, 1000 ml for samples diluted as described in Section 11.2.1. 10−3 = Conversion factor, µg/ng. S = Aliquot volume added to aeration cell, ml.12.4 Mercury Emission Rate. Calculate the daily Hg emission rate, R, using Equation 101-2. For continuous operations, the operating time is equal to 86,400 seconds per day. For cyclic operations, use only the time per day each stack is in operation. The total Hg emission rate from a source will be the summation of results from all stacks.
Where: K1 = 0.3858 °K/mm Hg for metric units. K1 = 17.64 °R/in. Hg for English units. K3 = 10−6 g/µg for metric units. = 2.2046 “ × 10−9 lb/µg for English units. Ps = Absolute stack gas pressure, mm Hg (in. Hg). t = Daily operating time, sec/day. Ts = Absolute average stack gas temperature, °K (°R). Vm(std) = Dry gas sample volume at standard conditions, scm (scf). Vw(std) = Volume of water vapor at standard conditions, scm (scf).12.5 Determination of Compliance. Each performance test consists of three repetitions of the applicable test method. For the purpose of determining compliance with an applicable national emission standard, use the average of the results of all repetitions.
13.0 Method PerformanceThe following estimates are based on collaborative tests, wherein 13 laboratories performed duplicate analyses on two Hg-containing samples from a chlor-alkali plant and on one laboratory-prepared sample of known Hg concentration. The sample concentrations ranged from 2 to 65 µg Hg/ml.
13.1 Precision. The estimated intra-laboratory and inter-laboratory standard deviations are 1.6 and 1.8 µg Hg/ml, respectively.
13.2 Accuracy. The participating laboratories that analyzed a 64.3 µg Hg/ml (in 0.1 M ICl) standard obtained a mean of 63.7 µg Hg/ml.
13.3 Analytical Range. After initial dilution, the range of this method is 0.5 to 120 µg Hg/ml. The upper limit can be extended by further dilution of the sample.
14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 Alternative Procedures16.1 Alternative Analyzer. Samples may also be analyzed by cold vapor atomic fluorescence spectrometry.
17.0 ReferencesSame as Method 5, Section 17.0, References 1-3, 5, and 6, with the addition of the following:
1. Determining Dust Concentration in a Gas Stream. ASME Performance Test Code No. 27. New York, NY. 1957.
2. DeVorkin, Howard, et al. Air Pollution Source Testing Manual. Air Pollution Control District. Los Angeles, CA. November 1963.
3. Hatch, W.R., and W.I. Ott. Determination of Sub-Microgram Quantities of Mercury by Atomic Absorption Spectrophotometry. Anal. Chem. 40:2085-87. 1968.
4. Mark, L.S. Mechanical Engineers' Handbook. McGraw-Hill Book Co., Inc. New York, NY. 1951.
5. Western Precipitation Division of Joy Manufacturing Co. Methods for Determination of Velocity, Volume, Dust and Mist Content of Gases. Bulletin WP-50. Los Angeles, CA. 1968.
6. Perry, J.H. Chemical Engineers' Handbook. McGraw-Hill Book Co., Inc. New York, NY. 1960.
7. Shigehara, R.T., W.F. Todd, and W.S. Smith. Significance of Errors in Stack Sampling Measurements. Stack Sampling News. 1(3):6-18. September 1973.
8. Smith, W.S., R.T. Shigehara, and W.F. Todd. A Method of Interpreting Stack Sampling Data. Stack Sampling News. 1(2):8-17. August 1973.
9. Standard Method for Sampling Stacks for Particulate Matter. In: 1971 Annual Book of ASTM Standards, Part 23. ASTM Designation D 2928-71. Philadelphia, PA 1971.
10. Vennard, J.K. Elementary Fluid Mechanics. John Wiley and Sons, Inc. New York. 1947.
11. Mitchell, W.J. and M.R. Midgett. Improved Procedure for Determining Mercury Emissions from Mercury Cell Chlor-Alkali Plants. J. APCA. 26:674-677. July 1976.
12. Shigehara, R.T. Adjustments in the EPA Nomograph for Different Pitot Tube Coefficients and Dry Molecular Weights. Stack Sampling News. 2:4-11. October 1974.
13. Vollaro, R.F. Recommended Procedure for Sample Traverses in Ducts Smaller than 12 Inches in Diameter. U.S. Environmental Protection Agency, Emission Measurement Branch. Research Triangle Park, NC. November 1976.
14. Klein, R. and C. Hach. Standard Additions: Uses and Limitation in Spectrophotometric Measurements. Amer. Lab. 9:21. 1977.
15. Perkin Elmer Corporation. Analytical Methods for Atomic Absorption Spectrophotometry. Norwalk, Connecticut. September 1976.
18.0 Tables, Diagrams, Flowcharts, and Validation Data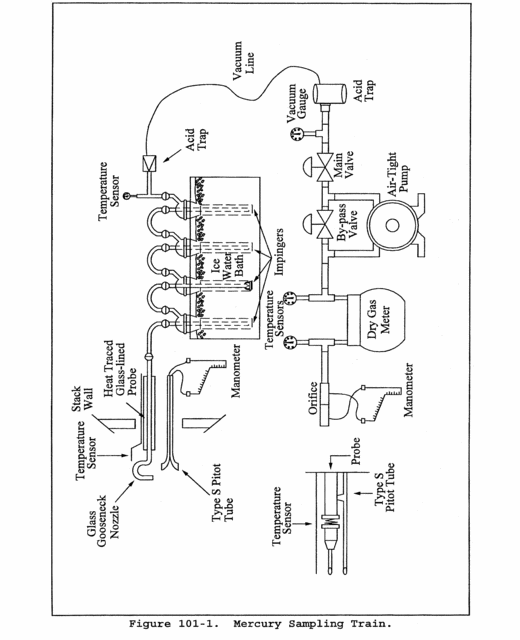
 Method 101A -
Determination of Particulate and Gaseous Mercury Emissions From
Sewage Sludge Incinerators Note:
Method 101A -
Determination of Particulate and Gaseous Mercury Emissions From
Sewage Sludge Incinerators Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from methods in appendix A to 40 CFR part 60 and in this part. Therefore, to obtain reliable results, persons using this method should also have a thorough knowledge of at least the following additional test methods: Methods 1, Method 2, Method 3, and Method 5 of part 60 (appendix A), and Method 101 part 61 (appendix B).
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Mercury (Hg) | 7439-97-6 | Dependent upon spectrophotometer and recorder. |
1.2 Applicability. This method is applicable for the determination of Hg emissions from sewage sludge incinerators and other sources as specified in an applicable subpart of the regulations.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method2.1 Particulate and gaseous Hg emissions are withdrawn isokinetically from the source and are collected in acidic potassium permanganate (KMnO4) solution. The Hg collected (in the mercuric form) is reduced to elemental Hg, which is then aerated from the solution into an optical cell and measured by atomic absorption spectrophotometry.
3.0 Definitions. [Reserved] 4.0 Interferences4.1 Sample Collection. Excessive oxidizable organic matter in the stack gas prematurely depletes the KMnO4 solution and thereby prevents further collection of Hg.
4.2 Analysis. Condensation of water vapor on the optical cell windows causes a positive interference.
5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrochloric Acid (HCl). Highly toxic. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Nitric Acid (HNO3). Highly corrosive to eyes, skin, nose, and lungs. Vapors cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.3 Sulfuric acid (H2SO4). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 3 mg/m 3 will cause lung damage in uninitiated. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
5.3 Chlorine Evolution. Hydrochloric acid reacts with KMnO4 to liberate chlorine gas. Although this is a minimal concern when small quantities of HCl (5-10 ml) are used in the impinger rinse, a potential safety hazard may still exist. At sources that emit higher concentrations of oxidizable materials (e.g., power plants), more HCl may be required to remove the larger amounts of brown deposit formed in the impingers. In such cases, the potential safety hazards due to sample container pressurization are greater, because of the larger volume of HCl rinse added to the recovered sample. These hazards are eliminated by storing and analyzing the HCl impinger wash separately from the permanganate impinger sample.
6.0 Equipment and Supplies6.1 Sample Collection and Sample Recovery. Same as Method 101, Sections 6.1 and 6.2, respectively, with the following exceptions:
6.1.1 Probe Liner. Same as in Method 101, Section 6.1.2, except that if a filter is used ahead of the impingers, the probe heating system must be used to minimize the condensation of gaseous Hg.
6.1.2 Filter Holder (Optional). Borosilicate glass with a rigid stainless-steel wire-screen filter support (do not use glass frit supports) and a silicone rubber or Teflon gasket, designed to provide a positive seal against leakage from outside or around the filter. The filter holder must be equipped with a filter heating system capable of maintaining a temperature around the filter holder of 120 ±14 °C (248 ±25 °F) during sampling to minimize both water and gaseous Hg condensation. A filter may also be used in cases where the stream contains large quantities of particulate matter.
6.2 Sample Analysis. Same as Method 101, Section 6.3, with the following additions and exceptions:
6.2.1 Volumetric Pipets. Class A; 1-, 2-, 3-, 4-, 5-, 10-, and 20-ml.
6.2.2 Graduated Cylinder. 25-ml.
6.2.3 Steam Bath.
6.2.4 Atomic Absorption Spectrophotometer or Equivalent. Any atomic absorption unit with an open sample presentation area in which to mount the optical cell is suitable. Instrument settings recommended by the particular manufacturer should be followed. Instruments designed specifically for the measurement of mercury using the cold-vapor technique are commercially available and may be substituted for the atomic absorption spectrophotometer.
6.2.5 Optical Cell. Alternatively, a heat lamp mounted above the cell or a moisture trap installed upstream of the cell may be used.
6.2.6 Aeration Cell. Alternatively, aeration cells available with commercial cold vapor instrumentation may be used.
6.2.7 Aeration Gas Cylinder. Nitrogen, argon, or dry, Hg-free air, equipped with a single-stage regulator. Alternatively, aeration may be provided by a peristaltic metering pump. If a commercial cold vapor instrument is used, follow the manufacturer's recommendations.
7.0 Reagents and StandardsUnless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection and Recovery. The following reagents are required for sample collection and recovery:
7.1.1 Water. Deionized distilled, to conform to ASTM D 1193-77 or 91 Type 1. If high concentrations of organic matter are not expected to be present, the analyst may eliminate the KMnO4 test for oxidizable organic matter. Use this water in all dilutions and solution preparations.
7.1.2 Nitric Acid, 50 Percent (V/V). Mix equal volumes of concentrated HNO3 and water, being careful to add the acid to the water slowly.
7.1.3 Silica Gel. Indicating type, 6 to 16 mesh. If previously used, dry at 175 °C (350 °F) for 2 hours. New silica gel may be used as received.
7.1.4 Filter (Optional). Glass fiber filter, without organic binder, exhibiting at least 99.95 percent efficiency on 0.3-μm dioctyl phthalate smoke particles. The filter in cases where the gas stream contains large quantities of particulate matter, but blank filters should be analyzed for Hg content.
7.1.5 Sulfuric Acid, 10 Percent (V/V). Carefully add and mix 100 ml of concentrated H2SO4 to 900 ml of water.
7.1.6 Absorbing Solution, 4 Percent KMnO4 (W/V). Prepare fresh daily. Dissolve 40 g of KMnO4 in sufficient 10 percent H2SO4 to make 1 liter. Prepare and store in glass bottles to prevent degradation.
7.1.7 Hydrochloric Acid, 8 N. Carefully add and mix 67 ml of concentrated HCl to 33 ml of water.
7.2 Sample Analysis. The following reagents and standards are required for sample analysis:
7.2.1 Water. Same as in Section 7.1.1.
7.2.2 Tin (II) Solution. Prepare fresh daily, and keep sealed when not being used. Completely dissolve 20 g of tin (II) chloride (or 25 g of tin (II) sulfate) crystals (Baker Analyzed reagent grade or any other brand that will give a clear solution) in 25 ml of concentrated HCl. Dilute to 250 ml with water. Do not substitute HNO3 H2SO4, or other strong acids for the HCl.
7.2.3 Sodium Chloride-Hydroxylamine Solution. Dissolve 12 g of sodium chloride and 12 g of hydroxylamine sulfate (or 12 g of hydroxylamine hydrochloride) in water and dilute to 100 ml.
7.2.4 Hydrochloric Acid, 8 N. Same as Section 7.1.7.
7.2.5 Nitric Acid, 15 Percent (V/V). Carefully add 15 ml HNO3 to 85 ml of water.
7.2.6 Antifoam B Silicon Emulsion. J.T. Baker Company (or equivalent).
7.2.7 Mercury Stock Solution, 1 mg Hg/ml. Prepare and store all Hg standard solutions in borosilicate glass containers. Completely dissolve 0.1354 g of Hg (II) chloride in 75 ml of water. Add 10 ml of concentrated HNO3, and adjust the volume to exactly 100 ml with water. Mix thoroughly. This solution is stable for at least one month.
7.2.8 Intermediate Hg Standard Solution, 10 µg/ml. Prepare fresh weekly. Pipet 5.0 ml of the Hg stock solution (Section 7.2.7) into a 500 ml volumetric flask, and add 20 ml of 15 percent HNO3 solution. Adjust the volume to exactly 500 ml with water. Thoroughly mix the solution.
7.2.9 Working Hg Standard Solution, 200 ng Hg/ml. Prepare fresh daily. Pipet 5.0 ml from the “Intermediate Hg Standard Solution” (Section 7.2.8) into a 250-ml volumetric flask. Add 5 ml of 4 percent KMnO4 absorbing solution and 5 ml of 15 percent HNO3. Adjust the volume to exactly 250 ml with water. Mix thoroughly.
7.2.10 Potassium Permanganate, 5 Percent (W/V). Dissolve 5 g of KMnO4 in water and dilute to 100 ml.
7.2.11 Filter. Whatman No. 40, or equivalent.
8.0 Sample Collection, Preservation, Transport, and StorageSame as Method 101, Section 8.0, with the exception of the following:
8.1 Preliminary Determinations. Same as Method 101, Section 8.2, except that the liberation of free iodine in the first impinger due to high Hg or sulfur dioxide concentrations is not applicable. In this method, high oxidizable organic content may make it impossible to sample for the desired minimum time. This problem is indicated by the complete bleaching of the purple color of the KMnO4 solution. In cases where an excess of water condensation is encountered, collect two runs to make one sample, or add an extra impinger in front of the first impinger (also containing acidified KMnO4 solution).
8.2 Preparation of Sampling Train. Same as Method 101, Section 8.3, with the exception of the following:
8.2.1 In this method, clean all the glass components by rinsing with 50 percent HNO3, tap water, 8 N HCl, tap water, and finally with deionized distilled water. Then place 50 ml of absorbing solution in the first impinger and 100 ml in each of the second and third impingers.
8.2.2 If a filter is used, use a pair of tweezers to place the filter in the filter holder. Be sure to center the filter, and place the gasket in the proper position to prevent the sample gas stream from bypassing the filter. Check the filter for tears after assembly is completed. Be sure also to set the filter heating system at the desired operating temperature after the sampling train has been assembled.
8.3 Sampling Train Operation. In addition to the procedure outlined in Method 101, Section 8.5, maintain a temperature around the filter (if applicable) of 120 ±14 °C (248 ±25 °F).
8.4 Sample Recovery. Same as Method 101, Section 8.7, with the exception of the following:
8.4.1 Transfer the probe, impinger assembly, and (if applicable) filter assembly to the cleanup area.
8.4.2 Treat the sample as follows:
8.4.2.1 Container No. 1 (Impinger, Probe, and Filter Holder) and, if applicable, Container No. 1A (HCl rinse).
8.4.2.1.1 Using a graduated cylinder, measure the liquid in the first three impingers to within 1 ml. Record the volume of liquid present (e.g., see Figure 5-6 of Method 5). This information is needed to calculate the moisture content of the effluent gas. (Use only graduated cylinder and glass storage bottles that have been precleaned as in Section 8.2.1.) Place the contents of the first three impingers (four if an extra impinger was added as described in Section 8.1) into a 1000-ml glass sample bottle labeled Container No. 1.
Note:If a filter is used, remove the filter from its holder as outlined under Section 8.4.3.
8.4.2.1.2 Taking care that dust on the outside of the probe or other exterior surfaces does not get into the sample, quantitatively recover the Hg (and any condensate) from the probe nozzle, probe fitting, probe liner, front half of the filter holder (if applicable), and impingers as follows: Rinse these components with a total of 400 ml (350 ml if an extra impinger was added as described in Section 8.1) of fresh absorbing solution, carefully assuring removal of all loose particulate matter from the impingers; add all washings to the 1000 ml glass sample bottle. To remove any residual brown deposits on the glassware following the permanganate rinse, rinse with approximately 100 ml of water, carefully assuring removal of all loose particulate matter from the impingers. Add this rinse to Container No. 1.
8.4.2.1.3 If no visible deposits remain after this water rinse, do not rinse with 8 N HCl. If deposits do remain on the glassware after the water rinse, wash impinger walls and stems with 25 ml of 8 N HCl, and place the wash in a separate container labeled Container No. 1A as follows: Place 200 ml of water in a sample container labeled Container No. 1A. Wash the impinger walls and stem with the HCl by turning the impinger on its side and rotating it so that the HCl contacts all inside surfaces. Pour the HCl wash carefully with stirring into Container No. 1A.
8.4.2.1.4 After all washings have been collected in the appropriate sample container(s), tighten the lid(s) on the container(s) to prevent leakage during shipment to the laboratory. Mark the height of the fluid level to allow subsequent determination of whether leakage has occurred during transport. Label each container to identify its contents clearly.
8.4.3 Container No. 2 (Silica Gel). Same as Method 5, Section 8.7.6.3.
8.4.4 Container No. 3 (Filter). If a filter was used, carefully remove it from the filter holder, place it in a 100-ml glass sample bottle, and add 20 to 40 ml of absorbing solution. If it is necessary to fold the filter, be sure that the particulate cake is inside the fold. Carefully transfer to the 100-ml sample bottle any particulate matter and filter fibers that adhere to the filter holder gasket by using a dry Nylon bristle brush and a sharp-edged blade. Seal the container. Label the container to identify its contents clearly. Mark the height of the fluid level to allow subsequent determination of whether leakage has occurred during transport.
8.4.5 Container No. 4 (Filter Blank). If a filter was used, treat an unused filter from the same filter lot as that used for sampling according to the procedures outlined in Section 8.4.4.
8.4.6 Container No. 5 (Absorbing Solution Blank). Place 650 ml of 4 percent KMnO4 absorbing solution in a 1000-ml sample bottle. Seal the container.
8.4.7 Container No. 6 (HCl Rinse Blank). Place 200 ml of water in a 1000-ml sample bottle, and add 25 ml of 8 N HCl carefully with stirring. Seal the container. Only one blank sample per 3 runs is required.
9.0 Quality Control9.1 Miscellaneous Quality Control Measures.
| Section | Quality control measure | Effect |
|---|---|---|
| 8.0, 10.0 | Sampling equipment leak-checks and calibration | Ensure accuracy and precision of sampling measurements. |
| 10.2 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
| 11.3.3 | Check for matrix effects | Eliminate matrix effects. |
9.2 Volume Metering System Checks. Same as Method 5, Section 9.2.
10.0 Calibration and StandardizationSame as Method 101, Section 10.0, with the following exceptions:
10.1 Optical Cell Heating System Calibration. Same as in Method 101, Section 10.4, except use a-25 ml graduated cylinder to add 25 ml of water to the bottle section of the aeration cell.
10.2 Spectrophotometer and Recorder Calibration.
10.2.1 The Hg response may be measured by either peak height or peak area.
Note:The temperature of the solution affects the rate at which elemental Hg is released from a solution and, consequently, it affects the shape of the absorption curve (area) and the point of maximum absorbance (peak height). To obtain reproducible results, all solutions must be brought to room temperature before use.
10.2.2 Set the spectrophotometer wave length at 253.7 nm, and make certain the optical cell is at the minimum temperature that will prevent water condensation. Then set the recorder scale as follows: Using a 25-ml graduated cylinder, add 25 ml of water to the aeration cell bottle. Add three drops of Antifoam B to the bottle, and then pipet 5.0 ml of the working Hg standard solution into the aeration cell.
Note:Always add the Hg-containing solution to the aeration cell after the 25 ml of water.
10.2.3 Place a Teflon-coated stirring bar in the bottle. Add 5 ml of absorbing solution to the aeration bottle, and mix well. Before attaching the bottle section to the bubbler section of the aeration cell, make certain that (1) the aeration cell exit arm stopcock (Figure 101-3 of Method 101) is closed (so that Hg will not prematurely enter the optical cell when the reducing agent is being added) and (2) there is no flow through the bubbler. If conditions (1) and (2) are met, attach the bottle section to the bubbler section of the aeration cell. Add sodium chloride-hydroxylamine in 1 ml increments until the solution is colorless. Now add 5 ml of tin (II) solution to the aeration bottle through the side arm, and immediately stopper the side arm. Stir the solution for 15 seconds, turn on the recorder, open the aeration cell exit arm stopcock, and immediately initiate aeration with continued stirring. Determine the maximum absorbance of the standard, and set this value to read 90 percent of the recorder full scale.
11.0 Analytical Procedure11.1 Sample Loss Check. Check the liquid level in each container to see if liquid was lost during transport. If a noticeable amount of leakage occurred, either void the sample or use methods subject to the approval of the Administrator to account for the losses.
11.2 Sample Preparation. Treat sample containers as follows:
11.2.1 Containers No. 3 and No. 4 (Filter and Filter Blank).
11.2.1.1 If a filter is used, place the contents, including the filter, of Containers No. 3 and No. 4 in separate 250-ml beakers, and heat the beakers on a steam bath until most of the liquid has evaporated. Do not heat to dryness. Add 20 ml of concentrated HNO3 to the beakers, cover them with a watch glass, and heat on a hot plate at 70 °C (160 °F) for 2 hours. Remove from the hot plate.
11.2.1.2 Filter the solution from digestion of the Container No. 3 contents through Whatman No. 40 filter paper, and save the filtrate for addition to the Container No. 1 filtrate as described in Section 11.2.2. Discard the filter paper.
11.2.1.3 Filter the solution from digestion of the Container No. 4 contents through Whatman No. 40 filter paper, and save the filtrate for addition to Container No. 5 filtrate as described in Section 11.2.3 below. Discard the filter paper.
11.2.2 Container No. 1 (Impingers, Probe, and Filter Holder) and, if applicable, No. 1A (HCl rinse).
11.2.2.1 Filter the contents of Container No. 1 through Whatman No. 40 filter paper into a 1 liter volumetric flask to remove the brown manganese dioxide (MnO2) precipitate. Save the filter for digestion of the brown MnO2 precipitate. Add the sample filtrate from Container No. 3 to the 1-liter volumetric flask, and dilute to volume with water. If the combined filtrates are greater than 1000 ml, determine the volume to the nearest ml and make the appropriate corrections for blank subtractions. Mix thoroughly. Mark the filtrate as analysis Sample No. A.1 and analyze for Hg within 48 hr of the filtration step. Place the saved filter, which was used to remove the brown MnO2 precipitate, into an appropriate sized container. In a laboratory hood, add 25 ml of 8 N HCl to the filter and allow to digest for a minimum of 24 hours at room temperature.
11.2.2.2 Filter the contents of Container 1A through Whatman No. 40 filter paper into a 500-ml volumetric flask. Then filter the digestate of the brown MnO2 precipitate from Container No. 1 through Whatman No. 40 filter paper into the same 500-ml volumetric flask, and dilute to volume with water. Mark this combined 500 ml dilute solution as analysis Sample No. A.2. Discard the filters.
11.2.3 Container No. 5 (Absorbing Solution Blank) and No. 6 (HCl Rinse Blank).
11.2.3.1 Treat Container No. 5 as Container No. 1 (as described in Section 11.2.2), except substitute the filter blank filtrate from Container No. 4 for the sample filtrate from Container No. 3, and mark as Sample A.1 Blank.
11.2.3.2 Treat Container No. 6 as Container No. 1A, (as described in Section 11.2.2, except substitute the filtrate from the digested blank MnO2 precipitate for the filtrate from the digested sample MnO2 precipitate, and mark as Sample No. A.2 Blank.
Note:When analyzing samples A.1 Blank and HCl A.2 Blank, always begin with 10 ml aliquots. This applies specifically to blank samples.
11.3 Analysis. Calibrate the analytical equipment and develop a calibration curve as outlined in Section 10.0.
11.3.1 Mercury Samples. Then repeat the procedure used to establish the calibration curve with appropriately sized aliquots (1 to 10 ml) of the samples (from Sections 11.2.2 and 11.2.3) until two consecutive peak heights agree within 3 percent of their average value. If the 10 ml sample is below the detectable limit, use a larger aliquot (up to 20 ml), but decrease the volume of water added to the aeration cell accordingly to prevent the solution volume from exceeding the capacity of the aeration bottle. If the peak maximum of a 1.0 ml aliquot is off scale, further dilute the original sample to bring the Hg concentration into the calibration range of the spectrophotometer. If the Hg content of the absorbing solution and filter blank is below the working range of the analytical method, use zero for the blank.
11.3.2 Run a blank and standard at least after every five samples to check the spectrophotometer calibration; recalibrate as necessary.
11.3.3 Check for Matrix Effects (optional). Same as Method 101, Section 11.3.3.
12.0 Data Analysis and Calculations Note:Carry out calculations, retaining at least one extra decimal significant figure beyond that of the acquired data. Round off figures only after the final calculation. Other forms of the equations may be used as long as they give equivalent results.
12.1 Nomenclature.
C(fltr)Hg = Total ng of Hg in aliquot of KMnO4 filtrate and HNO3 digestion of filter analyzed (aliquot of analysis Sample No. A.1). C(fltr blk)Hg = Total ng of Hg in aliquot of KMnO4 blank and HNO3 digestion of blank filter analyzed (aliquot of analysis Sample No. A.1 blank). C(HC1 blk)Hg = Total ng of Hg analyzed in aliquot of the 500-ml analysis Sample No. HCl A.2 blank. C(HCl)Hg = Total ng of Hg analyzed in the aliquot from the 500-ml analysis Sample No. HCl A.2. DF = Dilution factor for the HCl-digested Hg-containing solution, Analysis Sample No. “HCl A.2.” DFblk = Dilution factor for the HCl-digested Hg containing solution, Analysis Sample No. “HCl A.2 blank.” (Refer to sample No. “HCl A.2” dilution factor above.) m(fltr)Hg = Total blank corrected µg of Hg in KMnO4 filtrate and HNO3 digestion of filter sample. m(HCl)Hg = Total blank corrected µg of Hg in HCl rinse and HCl digestate of filter sample. mHg = Total blank corrected Hg content in each sample, µg. S = Aliquot volume of sample added to aeration cell, ml. Sblk = Aliquot volume of blank added to aeration cell, ml. Vf(blk) = Solution volume of blank sample, 1000 ml for samples diluted as described in Section 11.2.2. Vf(fltr) = Solution volume of original sample, normally 1000 ml for samples diluted as described in Section 11.2.2. Vf(HCl) = Solution volume of original sample, 500 ml for samples diluted as described in Section 11.2.1. 10−3 = Conversion factor, µg/ng.12.2 Average Dry Gas Meter Temperature and Average Orifice Pressure Drop, Dry Gas Volume, Volume of Water Vapor Condensed, Moisture Content, Isokinetic Variation, and Stack Gas Velocity and Volumetric Flow Rate. Same as Method 5, Sections 12.2 through 12.5, 12.11, and 12.12, respectively.
12.3 Total Mercury.
12.3.1 For each source sample, correct the average maximum absorbance of the two consecutive samples whose peak heights agree within 3 percent of their average for the contribution of the blank. Use the calibration curve and these corrected averages to determine the final total weight of Hg in ng in the aeration cell for each source sample.
12.3.2 Correct for any dilutions made to bring the sample into the working range of the spectrophotometer.
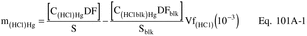 Note:
Note:
This dilution factor applies only to the intermediate dilution steps, since the original sample volume [(Vf)HCL] of “HCl A.2” has been factored out in the equation along with the sample aliquot (S). In Eq. 101A-1, the sample aliquot, S, is introduced directly into the aeration cell for analysis according to the procedure outlined in Section 11.3.1. A dilution factor is required only if it is necessary to bring the sample into the analytical instrument's calibration range.
Note:The maximum allowable blank subtraction for the HCl is the lesser of the two following values: (1) the actual blank measured value (analysis Sample No. HCl A.2 blank), or (2) 5% of the Hg content in the combined HCl rinse and digested sample (analysis Sample No. HCl A.2).
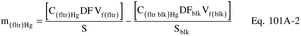 Note:
Note:
The maximum allowable blank subtraction for the HCl is the lesser of the two following values: (1) the actual blank measured value (analysis Sample No. “A.1 blank”), or (2) 5% of the Hg content in the filtrate (analysis Sample No. “A.1”).
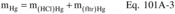
12.3 Mercury Emission Rate. Same as Method 101, Section 12.3.
12.4 Determination of Compliance. Same as Method 101, Section 12.4.
13.0 Method Performance13.1 Precision. Based on eight paired-train tests, the intra-laboratory standard deviation was estimated to be 4.8 µg/ml in the concentration range of 50 to 130 µg/m3.
13.2 Bias. [Reserved]
13.3 Range. After initial dilution, the range of this method is 20 to 800 ng Hg/ml. The upper limit can be extended by further dilution of the sample.
14.0 Pollution Prevention [Reserved] 15.0 Waste Management [Reserved] 16.0 Alternative Procedures16.1 Alternative Analyzers.
16.1.1 Inductively coupled plasma-atomic emission spectrometry (ICP-AES) may be used as an alternative to atomic absorption analysis provided the following conditions are met:
16.1.1.1 Sample collection, sample preparation, and analytical preparation procedures are as defined in the method except as necessary for the ICP-AES application.
16.1.1.2 The quality control procedures are conducted as prescribed.
16.1.1.3 The limit of quantitation for the ICP-AES must be demonstrated and the sample concentrations reported should be no less than two times the limit of quantitation. The limit of quantitation is defined as ten times the standard deviation of the blank value. The standard deviation of the blank value is determined from the analysis of seven blanks. It has been reported that for mercury and those elements that form hydrides, a continuous-flow generator coupled to an ICP-AES offers detection limits comparable to cold vapor atomic absorption.
16.1.2 Samples may also be analyzed by cold vapor atomic fluorescence spectrometry.
17.0 ReferencesSame as Section 16.0 of Method 101, with the addition of the following:
1. Mitchell, W.J., et al. Test Methods to Determine the Mercury Emissions from Sludge Incineration Plants. U.S. Environmental Protection Agency. Research Triangle Park, NC. Publication No. EPA-600/4-79-058. September 1979.
2. Wilshire, Frank W., et al. Reliability Study of the U.S. EPA's Method 101A - Determination of Particulate and Gaseous Mercury Emissions. U.S. Environmental Protection Agency. Research Triangle Park, NC. Report No. 600/D-31/219 AREAL 367, NTIS Acc No. PB91-233361.
3. Memorandum from William J. Mitchell to Roger T. Shigehara discussing the potential safety hazard in Section 7.2 of Method 101A. February 28, 1990.
18.0 Tables, Diagrams, Flowcharts, And Validation Data [Reserved] Method 102 - Determination of Particulate and Gaseous Mercury Emissions From Chlor-Alkali Plants (Hydrogen Streams) Note:This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part and in appendix A to 40 CFR part 60. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, Method 5, and Method 101.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Mercury (Hg) | 7439-97-6 | Dependent upon recorder and spectrophotometer. |
1.2 Applicability. This method is applicable for the determination of Hg emissions, including both particulate and gaseous Hg, from chlor-alkali plants and other sources (as specified in the regulations) where the carrier-gas stream in the duct or stack is principally hydrogen.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method2.1 Particulate and gaseous Hg emissions are withdrawn isokinetically from the source and collected in acidic iodine monochloride (ICl) solution. The Hg collected (in the mercuric form) is reduced to elemental Hg, which is then aerated from the solution into an optical cell and measured by atomic absorption spectrophotometry.
3.0 Definitions [Reserved] 4.0 InterferencesSame as Method 101, Section 4.2.
5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. Same as Method 101, Section 5.2.
5.3 Explosive Mixtures. The sampler must conduct the source test under conditions of utmost safety because hydrogen and air mixtures are explosive. Since the sampling train essentially is leakless, attention to safe operation can be concentrated at the inlet and outlet. If a leak does occur, however, remove the meter box cover to avoid a possible explosive mixture. The following specific precautions are recommended:
5.3.1 Operate only the vacuum pump during the test. The other electrical equipment, e.g., heaters, fans, and timers, normally are not essential to the success of a hydrogen stream test.
5.3.2 Seal the sample port to minimize leakage of hydrogen from the stack.
5.3.3 Vent sampled hydrogen at least 3 m (10 ft) away from the train. This can be accomplished by attaching a 13-mm (0.50-in.) ID Tygon tube to the exhaust from the orifice meter.
Note:A smaller ID tubing may cause the orifice meter calibration to be erroneous. Take care to ensure that the exhaust line is not bent or pinched.
6.0 Equipment and SuppliesSame as Method 101, Section 6.0, with the exception of the following:
6.1 Probe Heating System. Do not use, unless otherwise specified.
6.2 Glass Fiber Filter. Do not use, unless otherwise specified.
7.0 Reagents and StandardsSame as Method 101, Section 7.0.
8.0 Sample Collection, Preservation, Transport, and StorageSame as Method 101, Section 8.0, with the exception of the following:
8.1 Setting of Isokinetic Rates.
8.1.1 If a nomograph is used, take special care in the calculation of the molecular weight of the stack gas and in the setting of the nomograph to maintain isokinetic conditions during sampling (Sections 8.1.1.1 through 8.1.1.3 below).
8.1.1.1 Calibrate the meter box orifice. Use the techniques described in APTD-0576 (see Reference 9 in Section 17.0 of Method 5 of appendix A to part 60). Calibration of the orifice meter at flow conditions that simulate the conditions at the source is suggested. Calibration should either be done with hydrogen or with some other gas having a similar Reynolds Number so that there is similarity between the Reynolds Numbers during calibration and during sampling. Alternative mercury-free thermometers may be used if the thermometers are, at a minimum, equivalent in terms of performance or suitably effective for the specific temperature measurement application.
8.1.1.2 The nomograph described in APTD-0576 cannot be used to calculate the C factor because the nomograph is designed for use when the stack gas dry molecular weight is 29 ±4. Instead, the following calculation should be made to determine the proper C factor:
Where: Bws = Fraction by volume of water vapor in the stack gas. Cp = Pitot tube calibration coefficient, dimensionless. Md = Dry molecular weight of stack gas, lb/lb-mole. Ps = Absolute pressure of stack gas, in. Hg. Pm = Absolute pressure of gas at the meter, in. Hg. Tm = Absolute temperature of gas at the orifice, °R. ΔH@ = Meter box calibration factor obtained in Section 8.1.1.1, in. H2O. 0.00154 = (in. H2O/°R). Note:This calculation is left in English units, and is not converted to metric units because nomographs are based on English units.
8.1.1.3 Set the calculated C factor on the operating nomograph, and select the proper nozzle diameter and K factor as specified in APTD-0576. If the C factor obtained in Section 8.1.1.2 exceeds the values specified on the existing operating nomograph, expand the C scale logarithmically so that the values can be properly located.
8.1.2 If a calculator is used to set isokinetic rates, it is suggested that the isokinetic equation presented in Reference 13 in Section 17.0 of Method 101 be consulted.
8.2 Sampling in Small (<12-in. Diameter) Stacks. When the stack diameter (or equivalent diameter) is less than 12 inches, conventional pitot tube-probe assemblies should not be used. For sampling guidelines, see Reference 14 in Section 17.0 of Method 101.
9.0 Quality ControlSame as Method 101, Section 9.0.
10.0 Calibration and StandardizationsSame as Method 101, Section 10.0.
11.0 Analytical ProcedureSame as Method 101, Section 11.0.
12.0 Data Analysis and CalculationsSame as Method 101, Section 12.0.
13.0 Method PerformanceSame as Method 101, Section 13.0.
13.1 Analytical Range. After initial dilution, the range of this method is 0.5 to 120 µg Hg/ml. The upper limit can be extended by further dilution of the sample.
14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 ReferencesSame as Method 101, Section 16.0.
17.0 Tables, Diagrams, Flowcharts, and Validation Data. [Reserved] Method 103 - Beryllium Screening Method 1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Beryllium (Be) | 7440-41-7 | Dependent upon analytical procedure used. |
1.2 Applicability. This procedure details guidelines and requirements for methods acceptable for use in determining Be emissions in ducts or stacks at stationary sources.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method2.1 Particulate Be emissions are withdrawn isokinetically from three points in a duct or stack and are collected on a filter. The collected sample is analyzed for Be using an appropriate technique.
3.0 Definitions. [Reserved] 4.0 Interferences. [Reserved] 5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Hydrochloric Acid (HCl). Highly corrosive and toxic. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burn as thermal burn.
6.0 Equipment and Supplies6.1 Sample Collection. A schematic of the required sampling train configuration is shown in Figure 103-1 in Section 17.0. The essential components of the train are as follows:
6.1.1 Nozzle. Stainless steel, or equivalent, with sharp, tapered leading edge.
6.1.2 Probe. Sheathed borosilicate or quartz glass tubing.
6.1.3 Filter. Millipore AA, or equivalent, with appropriate filter holder that provides a positive seal against leakage from outside or around the filter. It is suggested that a Whatman 41, or equivalent, be placed immediately against the back side of the Millipore filter as a guard against breakage of the Millipore. Include the backup filter in the analysis. To be equivalent, other filters shall exhibit at least 99.95 percent efficiency (0.05 percent penetration) on 0.3 micron dioctyl phthalate smoke particles, and be amenable to the Be analysis procedure. The filter efficiency tests shall be conducted in accordance with ASTM D 2986-71, 78, 95a (incorporated by reference - see § 61.18). Test data from the supplier's quality control program are sufficient for this purpose.
6.1.4 Meter-Pump System. Any system that will maintain isokinetic sampling rate, determine sample volume, and is capable of a sampling rate of greater than 14 lpm (0.5 cfm).
6.2 Measurement of Stack Conditions. The following equipment is used to measure stack conditions:
6.2.1 Pitot Tube. Type S, or equivalent, with a constant coefficient (±5 percent) over the working range.
6.2.2 Inclined Manometer, or Equivalent. To measure velocity head to ±10 percent of the minimum value.
6.2.3 Temperature Measuring Device. To measure stack temperature to ±1.5 percent of the minimum absolute stack temperature.
6.2.4 Pressure Measuring Device. To measure stack pressure to ±2.5 mm Hg (0.1 in. Hg).
6.2.5 Barometer. To measure atmospheric pressure to ±2.5 mm Hg (0.1 in. Hg).
6.2.6 Wet and Dry Bulb Thermometers, Drying Tubes, Condensers, or Equivalent. To determine stack gas moisture content to ±1 percent.
6.3 Sample Recovery.
6.3.1 Probe Cleaning Equipment. Probe brush or cleaning rod at least as long as probe, or equivalent. Clean cotton balls, or equivalent, should be used with the rod.
6.3.2 Leakless Glass Sample Bottles. To contain sample.
6.4 Analysis. All equipment necessary to perform an atomic absorption, spectrographic, fluorometric, chromatographic, or equivalent analysis.
7.0 Reagents and Standards7.1 Sample Recovery.
7.1.1 Water. Deionized distilled, to conform to ASTM D 1193-77, 91 (incorporated by reference - see § 61.18), Type 3.
7.1.2 Acetone. Reagent grade.
7.1.3 Wash Acid, 50 Percent (V/V) Hydrochloric Acid (HCl). Mix equal volumes of concentrated HCl and water, being careful to add the acid slowly to the water.
7.2 Analysis. Reagents and standards as necessary for the selected analytical procedure.
8.0 Sample Collection, Preservation, Transport, and StorageGuidelines for source testing are detailed in the following sections. These guidelines are generally applicable; however, most sample sites differ to some degree and temporary alterations such as stack extensions or expansions often are required to insure the best possible sample site. Further, since Be is hazardous, care should be taken to minimize exposure. Finally, since the total quantity of Be to be collected is quite small, the test must be carefully conducted to prevent contamination or loss of sample.
8.1 Selection of a Sampling Site and Number of Sample Runs. Select a suitable sample site that is as close as practicable to the point of atmospheric emission. If possible, stacks smaller than one foot in diameter should not be sampled.
8.1.1 Ideal Sampling Site. The ideal sampling site is at least eight stack or duct diameters downstream and two diameters upstream from any flow disturbance such as a bend, expansion or contraction. For rectangular cross sections, use Equation 103-1 in Section 12.2 to determine an equivalent diameter, De.
8.1.2 Alternate Sampling Site. Some sampling situations may render the above sampling site criteria impractical. In such cases, select an alternate site no less than two diameters downstream and one-half diameter upstream from any point of flow disturbance. Additional sample runs are recommended at any sample site not meeting the criteria of Section 8.1.1.
8.1.3 Number of Sample Runs Per Test. Three sample runs constitute a test. Conduct each run at one of three different points. Select three points that proportionately divide the diameter, or are located at 25, 50, and 75 percent of the diameter from the inside wall. For horizontal ducts, sample on a vertical line through the centroid. For rectangular ducts, sample on a line through the centroid and parallel to a side. If additional sample runs are performed per Section 8.1.2, proportionately divide the duct to accommodate the total number of runs.
8.2 Measurement of Stack Conditions. Using the equipment described in Section 6.2, measure the stack gas pressure, moisture, and temperature to determine the molecular weight of the stack gas. Sound engineering estimates may be made in lieu of direct measurements. Describe the basis for such estimates in the test report.
8.3 Preparation of Sampling Train.
8.3.1 Assemble the sampling train as shown in Figure 103-1. It is recommended that all glassware be precleaned by soaking in wash acid for two hours.
8.3.2 Leak check the sampling train at the sampling site. The leakage rate should not be in excess of 1 percent of the desired sample rate.
8.4 Sampling Train Operation.
8.4.1 For each run, measure the velocity at the selected sampling point. Determine the isokinetic sampling rate. Record the velocity head and the required sampling rate. Place the nozzle at the sampling point with the tip pointing directly into the gas stream. Immediately start the pump and adjust the flow to isokinetic conditions. At the conclusion of the test, record the sampling rate. Again measure the velocity head at the sampling point. The required isokinetic rate at the end of the period should not have deviated more than 20 percent from that originally calculated. Describe the reason for any deviation beyond 20 percent in the test report.
8.4.2 Sample at a minimum rate of 14 liters/min (0.5 cfm). Obtain samples over such a period or periods of time as are necessary to determine the maximum emissions which would occur in a 24-hour period. In the case of cyclic operations, perform sufficient sample runs so as to allow determination or calculation of the emissions that occur over the duration of the cycle. A minimum sampling time of two hours per run is recommended.
8.5 Sample Recovery.
8.5.1 It is recommended that all glassware be precleaned as in Section 8.3. Sample recovery should also be performed in an area free of possible Be contamination. When the sampling train is moved, exercise care to prevent breakage and contamination. Set aside a portion of the acetone used in the sample recovery as a blank for analysis. The total amount of acetone used should be measured for accurate blank correction. Blanks can be eliminated if prior analysis shows negligible amounts.
8.5.2 Remove the filter (and backup filter, if used) and any loose particulate matter from filter holder, and place in a container.
8.5.3 Clean the probe with acetone and a brush or long rod and cotton balls. Wash into the container with the filter. Wash out the filter holder with acetone, and add to the same container.
9.0 Quality Control. [Reserved] 10.0 Calibration and Standardization10.1 Sampling Train. As a procedural check, compare the sampling rate regulation with a dry gas meter, spirometer, rotameter (calibrated for prevailing atmospheric conditions), or equivalent, attached to the nozzle inlet of the complete sampling train.
10.2 Analysis. Perform the analysis standardization as suggested by the manufacturer of the instrument, or the procedures for the analytical method in use.
11.0 Analytical ProcedureMake the necessary preparation of samples and analyze for Be. Any currently acceptable method (e.g., atomic absorption, spectrographic, fluorometric, chromatographic) may be used.
12.0 Data Analysis and Calculations12.1 Nomenclature.
As(avg) = Stack area, m 2 (ft 2). L = Length. R = Be emission rate, g/day. Vs(avg) = Average stack gas velocity, m/sec (ft/sec). Vtotal = Total volume of gas sampled, m 3 (ft 3). W = Width. Wt = Total weight of Be collected, mg. 10−6 = Conversion factor, g/µg. 86,400 = Conversion factor, sec/day.12.2 Calculate the equivalent diameter, De, for a rectangular cross section as follows:
12.3 Calculate the Be emission rate, R, in g/day for each stack using Equation 103-2. For cyclic operations, use only the time per day each stack is in operation. The total Be emission rate from a source is the summation of results from all stacks.
12.4 Test Report. Prepare a test report that includes as a minimum: A detailed description of the sampling train used, results of the procedural check described in Section 10.1 with all data and calculations made, all pertinent data taken during the test, the basis for any estimates made, isokinetic sampling calculations, and emission results. Include a description of the test site, with a block diagram and brief description of the process, location of the sample points in the stack cross section, and stack dimensions and distances from any point of disturbance.
13.0 Method Performance. [Reserved] 14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 References. [Reserved] 17.0 Tables, Diagrams, Flow Charts, and Validation Data Method
104 - Determination of Beryllium Emissions From Stationary Sources
Note:
Method
104 - Determination of Beryllium Emissions From Stationary Sources
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from methods in appendix A to 40 CFR part 60. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, and Method 5 in appendix A, part 60.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Beryllium (Be) | 7440-41-7 | Dependent upon recorder and spectrophotometer. |
1.2 Applicability. This method is applicable for the determination of Be emissions in ducts or stacks at stationary sources. Unless otherwise specified, this method is not intended to apply to gas streams other than those emitted directly to the atmosphere without further processing.
1.3 Data Quality Objectives. Adherences to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method2.1 Particulate and gaseous Be emissions are withdrawn isokinetically from the source and are collected on a glass fiber filter and in water. The collected sample is digested in an acid solution and is analyzed by atomic absorption spectrophotometry.
3.0 Definitions [Reserved] 4.0 Interferences4.1 Matrix Effects. Analysis for Be by flame atomic absorption spectrophotometry is sensitive to the chemical composition and to the physical properties (e.g., viscosity, pH) of the sample. Aluminum and silicon, in particular, are known to interfere when present in appreciable quantities. The analytical procedure includes (optionally) the use of the Method of Standard Additions to check for these matrix effects, and sample analysis using the Method of Standard Additions if significant matrix effects are found to be present (see Reference 2 in Section 17.0).
5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burn as thermal burn.
5.2.1 Hydrochloric Acid (HCl). Highly toxic. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Hydrogen Peroxide (H2O2). Irritating to eyes, skin, nose, and lungs.
5.2.3 Nitric Acid (HNO3). Highly corrosive to eyes, skin, nose, and lungs. Vapors cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.4 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.3 Beryllium is hazardous, and precautions should be taken to minimize exposure.
6.0 Equipment and Supplies6.1 Sample Collection. Same as Method 5, Section 6.1, with the exception of the following:
6.1.1 Sampling Train. Same as Method 5, Section 6.1.1, with the exception of the following:
6.1.2 Probe Liner. Borosilicate or quartz glass tubing. A heating system capable of maintaining a gas temperature of 120 ±14 °C (248 ±25 °F) at the probe exit during sampling to prevent water condensation may be used.
Note:Do not use metal probe liners.
6.1.3 Filter Holder. Borosilicate glass, with a glass frit filter support and a silicone rubber gasket. Other materials of construction (e.g., stainless steel, Teflon, Viton) may be used, subject to the approval of the Administrator. The holder design shall provide a positive seal against leakage from the outside or around the filter. The holder shall be attached immediately at the outlet of the probe. A heating system capable of maintaining the filter at a minimum temperature in the range of the stack temperature may be used to prevent condensation from occurring.
6.1.4 Impingers. Four Greenburg-Smith impingers connected in series with leak-free ground glass fittings or any similar leak-free noncontaminating fittings. For the first, third, and fourth impingers, use impingers that are modified by replacing the tip with a 13 mm-ID (0.5 in.) glass tube extending to 13 mm (0.5 in.) from the bottom of the flask may be used.
6.2 Sample Recovery. The following items are needed for sample recovery:
6.2.1 Probe Cleaning Rod. At least as long as probe.
6.2.2 Glass Sample Bottles. Leakless, with Teflon-lined caps, 1000 ml.
6.2.3 Petri Dishes. For filter samples, glass or polyethylene, unless otherwise specified by the Administrator.
6.2.4 Graduated Cylinder. 250 ml.
6.2.5 Funnel and Rubber Policeman. To aid in transfer of silica gel to container; not necessary if silica gel is weighed in the field.
6.2.6 Funnel. Glass, to aid in sample recovery.
6.2.7 Plastic Jar. Approximately 300 ml.
6.3 Analysis. The following items are needed for sample analysis:
6.3.1 Atomic Absorption Spectrophotometer. Perkin-Elmer 303, or equivalent, with nitrous oxide/acetylene burner.
6.3.2 Hot Plate.
6.3.3 Perchloric Acid Fume Hood.
7.0 Reagents and Standards Note:Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection. Same as Method 5, Section 7.1, including deionized distilled water conforming to ASTM D 1193-77 or 91 (incorporated by reference - see § 61.18), Type 3. The Millipore AA filter is recommended.
7.2 Sample Recovery. Same as Method 5 in appendix A, part 60, Section 7.2, with the addition of the following:
7.2.1 Wash Acid, 50 Percent (V/V) Hydrochloric Acid (HCl). Mix equal volumes of concentrated HCl and water, being careful to add the acid slowly to the water.
7.3 Sample Preparation and Analysis. The following reagents and standards and standards are needed for sample preparation and analysis:
7.3.1 Water. Same as in Section 7.1.
7.3.2. Perchloric Acid (HClO4). Concentrated (70 percent V/V).
7.3.3 Nitric Acid (HNO3). Concentrated.
7.3.4 Beryllium Powder. Minimum purity 98 percent.
7.3.5 Sulfuric Acid (H2SO4) Solution, 12 N. Dilute 33 ml of concentrated H2SO4 to 1 liter with water.
7.3.6 Hydrochloric Acid Solution, 25 Percent HCl (V/V).
7.3.7 Stock Beryllium Standard Solution, 10 µg Be/ml. Dissolve 10.0 mg of Be in 80 ml of 12 N H2SO4 in a 1000-ml volumetric flask. Dilute to volume with water. This solution is stable for at least one month. Equivalent strength Be stock solutions may be prepared from Be salts such as BeCl2 and Be(NO3)2 (98 percent minimum purity).
7.3.8 Working Beryllium Standard Solution, 1 µg Be/ml. Dilute a 10 ml aliquot of the stock beryllium standard solution to 100 ml with 25 percent HCl solution to give a concentration of 1 mg/ml. Prepare this dilute stock solution fresh daily.
8.0 Sample Collection, Preservation, Transport, and StorageThe amount of Be that is collected is generally small, therefore, it is necessary to exercise particular care to prevent contamination or loss of sample.
8.1 Pretest Preparation. Same as Method 5, Section 8.1, except omit Section 8.1.3.
8.2 Preliminary Determinations. Same as Method 5, Section 8.2, with the exception of the following:
8.2.1 Select a nozzle size based on the range of velocity heads to assure that it is not necessary to change the nozzle size in order to maintain isokinetic sampling rates below 28 liters/min (1.0 cfm).
8.2.2 Obtain samples over a period or periods of time that accurately determine the maximum emissions that occur in a 24-hour period. In the case of cyclic operations, perform sufficient sample runs for the accurate determination of the emissions that occur over the duration of the cycle. A minimum sample time of 2 hours per run is recommended.
8.3 Preparation of Sampling Train. Same as Method 5, Section 8.3, with the exception of the following:
8.3.1 Prior to assembly, clean all glassware (probe, impingers, and connectors) by first soaking in wash acid for 2 hours, followed by rinsing with water.
8.3.2 Save a portion of the water for a blank analysis.
8.3.3 Procedures relating to the use of metal probe liners are not applicable.
8.3.4 Probe and filter heating systems are needed only if water condensation is a problem. If this is the case, adjust the heaters to provide a temperature at or above the stack temperature. However, membrane filters such as the Millipore AA are limited to about 107 °C (225 °F). If the stack gas is in excess of about 93 °C (200 °F), consideration should be given to an alternate procedure such as moving the filter holder downstream of the first impinger to insure that the filter does not exceed its temperature limit. After the sampling train has been assembled, turn on and set the probe heating system, if applicable, at the desired operating temperature. Allow time for the temperatures to stabilize. Place crushed ice around the impingers.
Note:An empty impinger may be inserted between the third impinger and the silica gel to remove excess moisture from the sample stream.
8.4 Leak Check Procedures, Sampling Train Operation, and Calculation of Percent Isokinetic. Same as Method 5, Sections 8.4, 8.5, and 8.6, respectively.
8.5 Sample Recovery. Same as Method 5, Section 8.7, except treat the sample as follows: Transfer the probe and impinger assembly to a cleanup area that is clean, protected from the wind, and free of Be contamination. Inspect the train before and during this assembly, and note any abnormal conditions. Treat the sample as follows: Disconnect the probe from the impinger train.
8.5.1 Container No. 1. Same as Method 5, Section 8.7.6.1.
8.5.2 Container No. 2. Place the contents (measured to 1 ml) of the first three impingers into a glass sample bottle. Use the procedures outlined in Section 8.7.6.2 of Method 5, where applicable, to rinse the probe nozzle, probe fitting, probe liner, filter holder, and all glassware between the filter holder and the back half of the third impinger with water. Repeat this procedure with acetone. Place both water and acetone rinse solutions in the sample bottle with the contents of the impingers.
8.5.3 Container No. 3. Same as Method 5, Section 8.7.6.3.
8.6 Blanks.
8.6.1 Water Blank. Save a portion of the water as a blank. Take 200 ml directly from the wash bottle being used and place it in a plastic sample container labeled “H2O blank.”
8.6.2 Filter. Save two filters from each lot of filters used in sampling. Place these filters in a container labeled “filter blank.”
8.7 Post-test Glassware Rinsing. If an additional test is desired, the glassware can be carefully double rinsed with water and reassembled. However, if the glassware is out of use more than 2 days, repeat the initial acid wash procedure.
9.0 Quality Control| Section | Quality control measure | Effect |
|---|---|---|
| 8.4, 10.1 | Sampling equipment leak checks and calibration | Ensure accuracy and precision of sampling measurements. |
| 10.2 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
| 11.5 | Check for matrix effects | Eliminate matrix effects. |
Maintain a laboratory log of all calibrations.
10.1 Sampling Equipment. Same as Method 5, Section 10.0.
10.2 Preparation of Standard Solutions. Pipet 1, 3, 5, 8, and 10 ml of the 1.0 µg Be/ml working standard solution into separate 100 ml volumetric flasks, and dilute to the mark with water. The total amounts of Be in these standards are 1, 3, 5, 8, and 10 µg, respectively.
10.3 Spectrophotometer and Recorder. The Be response may be measured by either peak height or peak area. Analyze an aliquot of the 10-µg standard at 234.8 nm using a nitrous oxide/acetylene flame. Determine the maximum absorbance of the standard, and set this value to read 90 percent of the recorder full scale.
10.4 Calibration Curve.
10.4.1 After setting the recorder scale, analyze an appropriately sized aliquot of each standard and the BLANK (see Section 11) until two consecutive peaks agree within 3 percent of their average value.
10.4.3 Subtract the average peak height (or peak area) of the blank - which must be less than 2 percent of recorder full scale - from the averaged peak heights of the standards. If the blank absorbance is greater than 2 percent of full-scale, the probable cause is Be contamination of a reagent or carry-over of Be from a previous sample. Prepare the calibration curve by plotting the corrected peak height of each standard solution versus the corresponding total Be weight in the standard (in µg).
10.5 Spectrophotometer Calibration Quality Control. Calculate the least squares slope of the calibration curve. The line must pass through the origin or through a point no further from the origin than ±2 percent of the recorder full scale. Multiply the corrected peak height by the reciprocal of the least squares slope to determine the distance each calibration point lies from the theoretical calibration line. The difference between the calculated concentration values and the actual concentrations (i.e., 1, 3, 5, 8, and 10 µg Be) must be less than 7 percent for all standards.
11.0 Analytical Procedure11.1 Sample Loss Check. Prior to analysis, check the liquid level in Container No. 2. Note on the analytical data sheet whether leakage occurred during transport. If a noticeable amount of leakage occurred, either void the sample or take steps, subject to the approval of the Administrator, to adjust the final results.
11.2 Glassware Cleaning. Before use, clean all glassware according to the procedure of Section 8.3.1.
11.3 Sample Preparation. The digestion of Be samples is accomplished in part in concentrated HClO4.
Note:The sample must be heated to light brown fumes after the initial HNO3 addition; otherwise, dangerous perchlorates may result from the subsequent HClO4 digestion. HClO4 should be used only under a hood.
11.3.1 Container No. 1. Transfer the filter and any loose particulate matter from Container No. 1 to a 150-ml beaker. Add 35 ml concentrated HNO3. To oxidize all organic matter, heat on a hotplate until light brown fumes are evident. Cool to room temperature, and add 5 ml 12 N H2SO4 and 5 ml concentrated HClO4.
11.3.2 Container No. 2. Place a portion of the water and acetone sample into a 150 ml beaker, and put on a hotplate. Add portions of the remainder as evaporation proceeds and evaporate to dryness. Cool the residue, and add 35 ml concentrated HNO3. To oxidize all organic matter, heat on a hotplate until light brown fumes are evident. Cool to room temperature, and add 5 ml 12 N H2SO4 and 5 ml concentrated HClO4. Then proceed with step 11.3.4.
11.3.3 Final Sample Preparation. Add the sample from Section 11.3.2 to the 150-ml beaker from Section 11.3.1. Replace on a hotplate, and evaporate to dryness in a HClO4 hood. Cool the residue to room temperature, add 10.0 ml of 25 percent V/V HCl, and mix to dissolve the residue.
11.3.4 Filter and Water Blanks. Cut each filter into strips, and treat each filter individually as directed in Section 11.3.1. Treat the 200-ml water blank as directed in Section 11.3.2. Combine and treat these blanks as directed in Section 11.3.3.
11.4 Spectrophotometer Preparation. Turn on the power; set the wavelength, slit width, and lamp current; and adjust the background corrector as instructed by the manufacturer's manual for the particular atomic absorption spectrophotometer. Adjust the burner and flame characteristics as necessary.
11.5 Analysis. Calibrate the analytical equipment and develop a calibration curve as outlined in Sections 10.4 and 10.5.
11.5.1 Beryllium Samples. Repeat the procedure used to establish the calibration curve with an appropriately sized aliquot of each sample (from Section 11.3.3) until two consecutive peak heights agree within 3 percent of their average value. The peak height of each sample must be greater than 10 percent of the recorder full scale. If the peak height of the sample is off scale on the recorder, further dilute the original source sample to bring the Be concentration into the calibration range of the spectrophotometer.
11.5.2 Run a blank and standard at least after every five samples to check the spectrophotometer calibration. The peak height of the blank must pass through a point no further from the origin than ±2 percent of the recorder full scale. The difference between the measured concentration of the standard (the product of the corrected peak height and the reciprocal of the least squares slope) and the actual concentration of the standard must be less than 7 percent, or recalibration of the analyzer is required.
11.5.3 Check for Matrix Effects (optional). Use the Method of Standard Additions (see Reference 2 in Section 17.0) to check at least one sample from each source for matrix effects on the Be results. If the results of the Method of Standard Additions procedure used on the single source sample do not agree to within 5 percent of the value obtained by the routine atomic absorption analysis, then reanalyze all samples from the source using the Method of Standard Additions procedure.
11.6 Container No. 2 (Silica Gel). Weigh the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g using a balance. (This step may be conducted in the field.)
12.0 Data Analysis and CalculationsCarry out calculations, retaining at least one extra decimal significant figure beyond that of the acquired data. Round off figures only after the final calculation. Other forms of the equations may be used as long as they give equivalent results.
12.1 Nomenclature.
K1 = 0.3858 °K/mm Hg for metric units. = 17.64 °R/in. Hg for English units. K3 = 10−6 g/µg for metric units. = 2.2046 × 10−9 lb/µg for English units. mBe = Total weight of beryllium in the source sample. Ps = Absolute stack gas pressure, mm Hg (in. Hg). t = Daily operating time, sec/day. Ts = Absolute average stack gas temperature, °K (°R). Vm(std) = Dry gas sample volume at standard conditions, scm (scf). Vw(std) = Volume of water vapor at standard conditions, scm (scf).12.2 Average Dry Gas Meter Temperature and Average Orifice Pressure Drop, Dry Gas Volume, Volume of Water Vapor Condensed, Moisture Content, Isokinetic Variation, and Stack Gas Velocity and Volumetric Flow Rate. Same as Method 5, Sections 12.2 through 12.5, 12.11, and 12.12, respectively.
12.3 Total Beryllium. For each source sample, correct the average maximum absorbance of the two consecutive samples whose peak heights agree within 3 percent of their average for the contribution of the solution blank (see Sections 11.3.4 and 11.5.2). Correcting for any dilutions if necessary, use the calibration curve and these corrected averages to determine the total weight of Be in each source sample.
12.4 Beryllium Emission Rate. Calculate the daily Hg emission rate, R, using Equation 104-1. For continuous operations, the operating time is equal to 86,400 seconds per day. For cyclic operations, use only the time per day each stack is in operation. The total Hg emission rate from a source will be the summation of results from all stacks.
12.5 Determination of Compliance. Each performance test consists of three sample runs. For the purpose of determining compliance with an applicable national emission standard, use the average of the results of all sample runs.
13.0 Method Performance. [Reserved] 14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 Alternative Procedures16.1 Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES) Analysis. ICP-AES may be used as an alternative to atomic absorption analysis provided the following conditions are met:
16.1.1 Sample collection, sample preparation, and analytical preparation procedures are as defined in the method except as necessary for the ICP-AES application.
16.1.2 Quality Assurance/Quality Control procedures, including audit material analysis, are conducted as prescribed in the method. The QA acceptance conditions must be met.
16.1.3 The limit of quantitation for the ICP-AES must be demonstrated and the sample concentrations reported should be no less than two times the limit of quantitation. The limit of quantitation is defined as ten times the standard deviation of the blank value. The standard deviation of the blank value is determined from the analysis of seven blanks. It has been reported that for mercury and those elements that form hydrides, a continuous-flow generator coupled to an ICP-AES offers detection limits comparable to cold vapor atomic absorption.
16.2 Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) Analysis. ICP-MS may be used as an alternative to atomic absorption analysis.
16.3 Cold Vapor Atomic Fluorescence Spectrometry (CVAFS) Analysis. CVAFS may be used as an alternative to atomic absorption analysis.
17.0 ReferencesSame as References 1, 2, and 4-11 of Section 16.0 of Method 101 with the addition of the following:
1. Amos, M.D., and J.B. Willis. Use of High-Temperature Pre-Mixed Flames in Atomic Absorption Spectroscopy. Spectrochim. Acta. 22:1325. 1966.
2. Fleet, B., K.V. Liberty, and T. S. West. A Study of Some Matrix Effects in the Determination of Beryllium by Atomic Absorption Spectroscopy in the Nitrous Oxide-Acetylene Flame. Talanta 17:203. 1970.
18.0 Tables, Diagrams, Flowcharts, And Validation Data [Reserved] Method 105 - Determination of Mercury in Wastewater Treatment Plant Sewage Sludges Note:This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should also have a thorough knowledge of at least the following additional test methods: Method 101 and Method 101A.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Mercury (Hg) | 7439-97-6 | Dependent upon spectrophotometer and recorder. |
1.2 Applicability. This method is applicable for the determination of total organic and inorganic Hg content in sewage sludges.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method2.1 Time-composite sludge samples are withdrawn from the conveyor belt subsequent to dewatering and before incineration or drying. A weighed portion of the sludge is digested in aqua regia and is oxidized by potassium permanganate (KMnO4). Mercury in the digested sample is then measured by the conventional spectrophotometric cold-vapor technique.
3.0 Definitions [Reserved] 4.0 Interferences [Reserved] 5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burn as thermal burn.
5.2.1 Hydrochloric Acid (HCl). Highly toxic. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Nitric Acid (HNO3). Highly corrosive to eyes, skin, nose, and lungs. Vapors cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
6.0 Equipment and Supplies6.1 Sample Collection and Mixing. The following items are required for collection and mixing of the sludge samples:
6.1.1 Container. Plastic, 50-liter.
6.1.2 Scoop. To remove 950-ml (1 quart.) sludge sample.
6.1.3 Mixer. Mortar mixer, wheelbarrow-type, 57-liter (or equivalent) with electricity-driven motor.
6.1.4 Blender. Waring-type, 2-liter.
6.1.5 Scoop. To remove 100-ml and 20-ml samples of blended sludge.
6.1.6 Erlenmeyer Flasks. Four, 125-ml.
6.1.7 Beakers. Glass beakers in the following sizes: 50 ml (1), 200 ml (1), 400 ml (2).
6.2 Sample Preparation and Analysis. Same as Method 101, Section 6.3, with the addition of the following:
6.2.1 Hot Plate.
6.2.2 Desiccator.
6.2.3 Filter Paper. S and S No. 588 (or equivalent).
6.2.4 Beakers. Glass beakers, 200 ml and 400 ml (2 each).
7.0 Reagents and Standards Note:Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Analysis. Same as Method 101A, Section 7.2, with the following additions and exceptions:
7.1.1 Hydrochloric Acid. The concentrated HCl specified in Method 101A, Section 7.2.4, is not required.
7.1.2 Aqua Regia. Prepare immediately before use. Carefully add one volume of concentrated HNO3 to three volumes of concentrated HCl.
8.0 Sample Collection, Preservation, Storage, and Transport8.1 Sludge Sampling. Withdraw equal volume increments of sludge [for a total of at least 15 liters (16 quarts)] at intervals of 30 min over an 8-hr period, and combine in a rigid plastic container.
8.2 Sludge Mixing. Transfer the entire 15-liter sample to a mortar mixer. Mix the sample for a minimum of 30 min at 30 rpm. Take six 100-ml portions of sludge, and combine in a 2-liter blender. Blend sludge for 5 min; add water as necessary to give a fluid consistency. Immediately after stopping the blender, withdraw four 20-ml portions of blended sludge, and place them in separate, tared 125-ml Erlenmeyer flasks. Reweigh each flask to determine the exact amount of sludge added.
8.3 Sample Holding Time. Samples shall be analyzed within the time specified in the applicable subpart of the regulations.
9.0 Quality Control| Section | Quality control measure | Effect |
|---|---|---|
| 10.0 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
| 11.0 | Check for matrix effects | Eliminate matrix effects. |
Same as Method 101A, Section 10.2.
11.0 Analytical Procedures11.1 Solids Content of Blended Sludge. Dry one of the 20-ml blended samples from Section 8.2 in an oven at 105 °C (221 °F) to constant weight. Cool in a desiccator, weigh and record the dry weight of the sample.
11.2 Aqua Regia Digestion of Blended Samples.
11.2.1 To each of the three remaining 20-ml samples from Section 8.2 add 25 ml of aqua regia, and digest the on a hot plate at low heat (do not boil) for 30 min, or until samples are a pale yellow-brown color and are void of the dark brown color characteristic of organic matter. Remove from hotplate and allow to cool.
11.2.2 Filter each digested sample separately through an S and S No. 588 filter or equivalent, and rinse the filter contents with 50 ml of water. Transfer the filtrate and filter washing to a 100-ml volumetric flask, and carefully dilute to volume with water.
11.3 Solids Content of the Sludge Before Blending. Remove two 100-ml portions of mixed sludge from the mortar mixer and place in separate, tared 400-ml beakers. Reweigh each beaker to determine the exact amount of sludge added. Dry in oven at 105 °C (221 °F) and cool in a desiccator to constant weight.
11.4 Analysis for Mercury. Analyze the three aqua regia-digested samples using the procedures outlined in Method 101A, Section 11.0.
12.0 Data Analysis and Calculations12.1 Nomenclature.
Cm = Concentration of Hg in the digested sample, µg/g. Fsb = Weight fraction of solids in the blended sludge. Fsm = Weight fraction of solids in the collected sludge after mixing. M = Hg content of the sewage sludge (on a dry basis), µg/g. m = Mass of Hg in the aliquot of digested sample analyzed, µg. n = number of digested samples (specified in Section 11.2 as three). Va = Volume of digested sample analyzed, ml. Vs = Volume of digested sample, ml. Wb = Weight of empty sample beaker, g. Wbs = Weight of sample beaker and sample, g. Wbd = Weight of sample beaker and sample after drying, g. Wf = Weight of empty sample flask, g. Wfd = Weight of sample flask and sample after drying, g. Wfs = Weight of sample flask and sample, g.12.2 Mercury Content of Digested Sample (Wet Basis).
12.2.1 For each sample analyzed for Hg content, calculate the arithmetic mean maximum absorbance of the two consecutive samples whose peak heights agree ±3 percent of their average. Correct this average value for the contribution of the blank. Use the calibration curve and these corrected averages to determine the final Hg concentration in the solution cell for each sludge sample.
12.2.2 Calculate the average Hg concentration of the digested samples by correcting for any dilutions made to bring the sample into the working range of the spectrophotometer and for the weight of the sludge portion digested, using Equation 105-1.
12.3 Solids Content of Blended Sludge. Determine the solids content of the blended sludge using Equation 105-2.
12.4 Solids Content of Bulk Sample (before blending but, after mixing in mortar mixer). Determine the solids content of each 100 ml aliquot (Section 11.3), and average the results.
12.5 Mercury Content of Bulk Sample (Dry Basis). Average the results from the three samples from each 8-hr composite sample, and calculate the Hg concentration of the composite sample on a dry basis.
13.0 Method Performance13.1 Range. The range of this method is 0.2 to 5 micrograms per gram; it may be extended by increasing or decreasing sample size.
14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 References1. Bishop, J.N. Mercury in Sediments. Ontario Water Resources Commission. Toronto, Ontario, Canada. 1971.
2. Salma, M. Private Communication. EPA California/Nevada Basin Office. Alameda, California.
3. Hatch, W.R. and W.L. Ott. Determination of Sub-Microgram Quantities of Mercury by Atomic Absorption Spectrophotometry. Analytical Chemistry. 40:2085. 1968.
4. Bradenberger, H., and H. Bader. The Determination of Nanogram Levels of Mercury in Solution by a Flameless Atomic Absorption Technique. Atomic Absorption Newsletter. 6:101. 1967.
5. Analytical Quality Control Laboratory (AQCL). Mercury in Sediment (Cold Vapor Technique) (Provisional Method). U.S. Environmental Protection Agency. Cincinnati, Ohio. April 1972.
6. Kopp, J.F., M.C. Longbottom, and L.B. Lobring. “Cold Vapor” Method for Determining Mercury. Journal AWWA. 64(1):20-25. 1972.
7. Manual of Methods for Chemical Analysis of Water and Wastes. U.S. Environmental Protection Agency. Cincinnati, Ohio. Publication No. EPA-624/2-74-003. December 1974. pp. 118-138.
8. Mitchell, W.J., M.R. Midgett, J. Suggs, R.J. Velton, and D. Albrink. Sampling and Homogenizing Sewage for Analysis. Environmental Monitoring and Support Laboratory, Office of Research and Development, U.S. Environmental Protection Agency. Research Triangle Park, N.C. March 1979. p. 7.
17.0 Tables, Diagrams, Flowcharts, and Validation Data. [Reserved] Method 106 - Determination of Vinyl Chloride Emissions From Stationary Sources 1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Vinyl Chloride (CH2:CHCl) | 75-01-4 | Dependent upon analytical equipment. |
1.2 Applicability. This method is applicable for the determination of vinyl chloride emissions from ethylene dichloride, vinyl chloride, and polyvinyl chloride manufacturing processes. This method does not measure vinyl chloride contained in particulate matter.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method2.1 An integrated bag sample of stack gas containing vinyl chloride is subjected to GC analysis using a flame ionization detector (FID).
3.0 Definitions. [Reserved] 4.0 Interferences4.1 Resolution interferences of vinyl chloride may be encountered on some sources. Therefore, the chromatograph operator should select the column and operating parameters best suited to the particular analysis requirements. The selection made is subject to approval of the Administrator. Approval is automatic, provided that confirming data are produced through an adequate supplemental analytical technique, and that the data are available for review by the Administrator. An example of this would be analysis with a different column or GC/mass spectroscopy.
5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Toxic Analyte. Care must be exercised to prevent exposure of sampling personnel to vinyl chloride, which is a carcinogen.
6.0 Equipment and Supplies6.1 Sample Collection (see Figure 106-1). The sampling train consists of the following components:
6.1.1 Probe. Stainless steel, borosilicate glass, Teflon tubing (as stack temperature permits), or equivalent, equipped with a glass wool plug to remove particulate matter.
6.1.2 Sample Lines. Teflon, 6.4-mm outside diameter, of sufficient length to connect probe to bag. Use a new unused piece for each series of bag samples that constitutes an emission test, and discard upon completion of the test.
6.1.3 Quick Connects. Stainless steel, male (2) and female (2), with ball checks (one pair without), located as shown in Figure 106-1.
6.1.4 Tedlar Bags. 50- to 100-liter capacity, to contain sample. Aluminized Mylar bags may be used if the samples are analyzed within 24 hours of collection.
6.1.5 Bag Containers. Rigid leak-proof containers for sample bags, with covering to protect contents from sunlight.
6.1.6 Needle Valve. To adjust sample flow rates.
6.1.7 Pump. Leak-free, with minimum of 2-liter/min capacity.
6.1.8 Charcoal Tube. To prevent admission of vinyl chloride and other organics to the atmosphere in the vicinity of samplers.
6.1.9 Flowmeter. For observing sampling flow rate; capable of measuring a flow range from 0.10 to 1.00 liter/min.
6.1.10 Connecting Tubing. Teflon, 6.4-mm outside diameter, to assemble sampling train (Figure 106-1).
6.1.11 Tubing Fittings and Connectors. Teflon or stainless steel, to assemble sampling training.
6.2 Sample Recovery. Teflon tubing, 6.4-mm outside diameter, to connect bag to GC sample loop. Use a new unused piece for each series of bag samples that constitutes an emission test, and discard upon conclusion of analysis of those bags.
6.3 Analysis. The following equipment is required:
6.3.1 Gas Chromatograph. With FID potentiometric strip chart recorder and 1.0 to 5.0-ml heated sampling loop in automatic sample valve. The chromatographic system shall be capable of producing a response to 0.1-ppmv vinyl chloride that is at least as great as the average noise level. (Response is measured from the average value of the base line to the maximum of the wave form, while standard operating conditions are in use.)
6.3.2 Chromatographic Columns. Columns as listed below. Other columns may be used provided that the precision and accuracy of the analysis of vinyl chloride standards are not impaired and that information is available for review confirming that there is adequate resolution of vinyl chloride peak. (Adequate resolution is defined as an area overlap of not more than 10 percent of the vinyl chloride peak by an interferent peak. Calculation of area overlap is explained in Procedure 1 of appendix C to this part: “Determination of Adequate Chromatographic Peak Resolution.”)
6.3.2.1 Column A. Stainless steel, 2.0 m by 3.2 mm, containing 80/100-mesh Chromasorb 102.
6.3.2.2 Column B. Stainless steel, 2.0 m by 3.2 mm, containing 20 percent GE SF-96 on 60/ip-mesh Chromasorb P AW; or stainless steel, 1.0 m by 3.2 mm containing 80/100-mesh Porapak T. Column B is required as a secondary column if acetaldehyde is present. If used, column B is placed after column A. The combined columns should be operated at 120 °C (250 °F).
6.3.3 Rate Meters (2). Rotameter , or equivalent, 100-ml/min capacity, with flow control valves.
6.3.4 Gas Regulators. For required gas cylinders.
6.3.5 Temperature Sensor. Accurate to ±1 °C (±2 °F), to measure temperature of heated sample loop at time of sample injection.
6.3.6 Barometer. Accurate to ±5 mm Hg, to measure atmospheric pressure around GC during sample analysis.
6.3.7 Pump. Leak-free, with minimum of 100-ml/min capacity.
6.3.8 Recorder. Strip chart type, optionally equipped with either disc or electronic integrator.
6.3.9 Planimeter. Optional, in place of disc or electronic integrator on recorder, to measure chromatograph peak areas.
6.4 Calibration and Standardization.
6.4.1 Tubing. Teflon, 6.4-mm outside diameter, separate pieces marked for each calibration concentration.
Note:The following items are required only if the optional standard gas preparation procedures (Section 10.1) are followed.
6.4.2 Tedlar Bags. Sixteen-inch-square size, with valve; separate bag marked for each calibration concentration.
6.4.3 Syringes. 0.5-ml and 50-µl, gas tight, individually calibrated to dispense gaseous vinyl chloride.
6.4.4 Dry Gas Meter with Temperature and Pressure Gauges. Singer Model DTM-115 with 802 index, or equivalent, to meter nitrogen in preparation of standard gas mixtures, calibrated at the flow rate used to prepare standards.
7.0 Reagents and Standards7.1 Analysis. The following reagents are required for analysis.
7.1.1 Helium or Nitrogen. Purity 99.9995 percent or greater, for chromatographic carrier gas.
7.1.2 Hydrogen. Purity 99.9995 percent or greater.
7.1.3 Oxygen or Air. Either oxygen (purity 99.99 percent or greater) or air (less than 0.1 ppmv total hydrocarbon content), as required by detector.
7.2 Calibration. Use one of the following options: either Sections 7.2.1 and 7.2.2, or Section 7.2.3.
7.2.1 Vinyl Chloride. Pure vinyl chloride gas certified by the manufacturer to contain a minimum of 99.9 percent vinyl chloride. If the gas manufacturer maintains a bulk cylinder supply of 99.9 + percent vinyl chloride, the certification analysis may have been performed on this supply, rather than on each gas cylinder prepared from this bulk supply. The date of gas cylinder preparation and the certified analysis must have been affixed to the cylinder before shipment from the gas manufacturer to the buyer.
7.2.2 Nitrogen. Same as described in Section 7.1.1.
7.2.3 Cylinder Standards. Gas mixture standards (50-,10-, and 5 ppmv vinyl chloride) in nitrogen cylinders may be used to directly prepare a chromatograph calibration curve as described in Section 10.3 if the following conditions are met: (a) The manufacturer certifies the gas composition with an accuracy of ±3 percent or better. (b) The manufacturer recommends a maximum shelf life over which the gas concentration does not change by greater than ±5 percent from the certified value. (c) The manufacturer affixes the date of gas cylinder preparation, certified vinyl chloride concentration, and recommended maximum shelf to the cylinder before shipment to the buyer.
7.2.3.1 Cylinder Standards Certification. The manufacturer shall certify the concentration of vinyl chloride in nitrogen in each cylinder by (a) directly analyzing each cylinder and (b) calibrating his analytical procedure on the day of cylinder analysis. To calibrate his analytical procedure, the manufacturer shall use as a minimum, a three point calibration curve. It is recommended that the manufacturer maintain (1) a high concentration calibration standard (between 50 and 100 ppmv) to prepare his calibration curve by an appropriate dilution technique and (2) a low-concentration calibration standard (between 5 and 10 ppmv) to verify the dilution technique used. If the difference between the apparent concentration read from the calibration curve and the true concentration assigned to the low-concentration calibration standard exceeds 5 percent of the true concentration, the manufacturer shall determine the source of error and correct it, then repeat the three-point calibration.
7.2.3.2 Verification of Manufacturer's Calibration Standards. Before using a standard, the manufacturer shall verify each calibration standard (a) by comparing it to gas mixtures prepared (with 99 mole percent vinyl chloride) in accordance with the procedure described in Section 7.2.1 or (b) calibrating it against vinyl chloride cylinder Standard Reference Materials (SRM's) prepared by the National Institute of Standards and Technology, if such SRM's are available. The agreement between the initially determined concentration value and the verification concentration value must be ±5 percent. The manufacturer must reverify all calibration standards on a time interval consistent with the shelf life of the cylinder standards sold.
8.0 Sample Collection, Preservation, Storage, and Transport Note:Performance of this method should not be attempted by persons unfamiliar with the operation of a gas chromatograph (GC) nor by those who are unfamiliar with source sampling, because knowledge beyond the scope of this presentation is required.
8.1 Bag Leak-Check. The following leak-check procedure is recommended, but not required, prior to sample collection. The post-test leak-check procedure is mandatory. Connect a water manometer and pressurize the bag to 5 to 10 cm H2O (2 to 4 in. H2O). Allow to stand for 10 min. Any displacement in the water manometer indicates a leak. Also, check the rigid container for leaks in this manner.
Note:An alternative leak-check method is to pressurize the bag to 5 to 10 cm H2O and allow it to stand overnight. A deflated bag indicates a leak. For each sample bag in its rigid container, place a rotameter in line between the bag and the pump inlet. Evacuate the bag. Failure of the rotameter to register zero flow when the bag appears to be empty indicates a leak.
8.2 Sample Collection. Assemble the sample train as shown in Figure 106-1. Join the quick connects as illustrated, and determine that all connection between the bag and the probe are tight. Place the end of the probe at the centroid of the stack and start the pump with the needle valve adjusted to yield a flow that will fill over 50 percent of bag volume in the specific sample period. After allowing sufficient time to purge the line several times, change the vacuum line from the container to the bag and evacuate the bag until the rotameter indicates no flow. Then reposition the sample and vacuum lines and begin the actual sampling, keeping the rate proportional to the stack velocity. At all times, direct the gas exiting the rotameter away from sampling personnel. At the end of the sample period, shut off the pump, disconnect the sample line from the bag, and disconnect the vacuum line from the bag container. Protect the bag container from sunlight.
8.3 Sample Storage. Keep the sample bags out of direct sunlight. When at all possible, analysis is to be performed within 24 hours, but in no case in excess of 72 hours of sample collection. Aluminized Mylar bag samples must be analyzed within 24 hours.
8.4 Post-test Bag Leak-Check. Subsequent to recovery and analysis of the sample, leak-check the sample bag according to the procedure outlined in Section 8.1.
9.0 Quality Control| Section | Quality control measure | Effect |
|---|---|---|
| 10.3 | Chromatograph calibration | Ensure precision and accuracy of chromatograph. |
Maintain a laboratory log of all calibrations.
10.1 Preparation of Vinyl Chloride Standard Gas Mixtures. (Optional Procedure-delete if cylinder standards are used.) Evacuate a 16-inch square Tedlar bag that has passed a leak-check (described in Section 8.1) and meter in 5.0 liters of nitrogen. While the bag is filling, use the 0.5-ml syringe to inject 250 µl of 99.9 + percent vinyl chloride gas through the wall of the bag. Upon withdrawing the syringe, immediately cover the resulting hole with a piece of adhesive tape. The bag now contains a vinyl chloride concentration of 50 ppmv. In a like manner use the 50 µl syringe to prepare gas mixtures having 10-and 5-ppmv vinyl chloride concentrations. Place each bag on a smooth surface and alternately depress opposite sides of the bag 50 times to further mix the gases. These gas mixture standards may be used for 10 days from the date of preparation, after which time new gas mixtures must be prepared. (Caution: Contamination may be a problem when a bag is reused if the new gas mixture standard is a lower concentration than the previous gas mixture standard.)
10.2 Determination of Vinyl Chloride Retention Time. (This section can be performed simultaneously with Section 10.3.) Establish chromatograph conditions identical with those in Section 11.3. Determine proper attenuator position. Flush the sampling loop with helium or nitrogen and activate the sample valve. Record the injection time, sample loop temperature, column temperature, carrier gas flow rate, chart speed, and attenuator setting. Record peaks and detector responses that occur in the absence of vinyl chloride. Maintain conditions with the equipment plumbing arranged identically to Section 11.2, and flush the sample loop for 30 seconds at the rate of 100 ml/min with one of the vinyl chloride calibration mixtures. Then activate the sample valve. Record the injection time. Select the peak that corresponds to vinyl chloride. Measure the distance on the chart from the injection time to the time at which the peak maximum occurs. This quantity divided by the chart speed is defined as the retention time. Since other organics may be present in the sample, positive identification of the vinyl chloride peak must be made.
10.3 Preparation of Chromatograph Calibration Curve. Make a GC measurement of each gas mixture standard (described in Section 7.2.3 or 10.1) using conditions identical to those listed in Sections 11.2 and 11.3. Flush the sampling loop for 30 seconds at the rate of 100 ml/min with one of the standard mixtures, and activate the sample valve. Record the concentration of vinyl chloride injected (Cc), attenuator setting, chart speed, peak area, sample loop temperature, column temperature, carrier gas flow rate, and retention time. Record the barometric pressure. Calculate Ac, the peak area multiplied by the attenuator setting. Repeat until two consecutive injection areas are within 5 percent, then plot the average of those two values versus Cc. When the other standard gas mixtures have been similarly analyzed and plotted, draw a straight line through the points derived by the least squares method. Perform calibration daily, or before and after the analysis of each emission test set of bag samples, whichever is more frequent. For each group of sample analyses, use the average of the two calibration curves which bracket that group to determine the respective sample concentrations. If the two calibration curves differ by more than 5 percent from their mean value, then report the final results by both calibration curves.
11.0 Analytical Procedure11.2 Sample Recovery. With a new piece of Teflon tubing identified for that bag, connect a bag inlet valve to the gas chromatograph sample valve. Switch the valve to receive gas from the bag through the sample loop. Arrange the equipment so the sample gas passes from the sample valve to 100-ml/min rotameter with flow control valve followed by a charcoal tube and a 1-in. H2O pressure gauge. Maintain the sample flow either by a vacuum pump or container pressurization if the collection bag remains in the rigid container. After sample loop purging is ceased, allow the pressure gauge to return to zero before activating the gas sampling valve.
11.3 Analysis.
11.3.1 Set the column temperature to 100 °C (210 °F) and the detector temperature to 150 °C (300 °F). When optimum hydrogen and oxygen (or air) flow rates have been determined, verify and maintain these flow rates during all chromatography operations. Using helium or nitrogen as the carrier gas, establish a flow rate in the range consistent with the manufacturer's requirements for satisfactory detector operation. A flow rate of approximately 40 ml/min should produce adequate separations. Observe the base line periodically and determine that the noise level has stabilized and that base line drift has ceased. Purge the sample loop for 30 seconds at the rate of 100 ml/min, shut off flow, allow the sample loop pressure to reach atmospheric pressure as indicated by the H2O manometer, then activate the sample valve. Record the injection time (the position of the pen on the chart at the time of sample injection), sample number, sample loop temperature, column temperature, carrier gas flow rate, chart speed, and attenuator setting. Record the barometric pressure. From the chart, note the peak having the retention time corresponding to vinyl chloride as determined in Section 10.2. Measure the vinyl chloride peak area, Am, by use of a disc integrator, electronic integrator, or a planimeter. Measure and record the peak heights, Hm. Record Am and retention time. Repeat the injection at least two times or until two consecutive values for the total area of the vinyl chloride peak agree within 5 percent of their average. Use the average value for these two total areas to compute the bag concentration.
11.3.2 Compare the ratio of Hm to Am for the vinyl chloride sample with the same ratio for the standard peak that is closest in height. If these ratios differ by more than 10 percent, the vinyl chloride peak may not be pure (possibly acetaldehyde is present) and the secondary column should be employed (see Section 6.3.2.2).
11.4 Determination of Bag Water Vapor Content. Measure the ambient temperature and barometric pressure near the bag. From a water saturation vapor pressure table, determine and record the water vapor content of the bag, Bwb, as a decimal figure. (Assume the relative humidity to be 100 percent unless a lesser value is known.)
12.0 Calculations and Data Analysis12.1 Nomenclature.
Am = Measured peak area. Af = Attenuation factor. Bwb = Water vapor content of the bag sample, as analyzed, volume fraction. Cb = Concentration of vinyl chloride in the bag, ppmv. Cc = Concentration of vinyl chloride in the standard sample, ppmv. Pi = Laboratory pressure at time of analysis, mm Hg. Pr = Reference pressure, the laboratory pressure recorded during calibration, mm Hg. Ti = Absolute sample loop temperature at the time of analysis, °K (°R). Tr = Reference temperature, the sample loop temperature recorded during calibration, °K (°R).12.2 Sample Peak Area. Determine the sample peak area, Ac, as follows:
12.3 Vinyl Chloride Concentration. From the calibration curves prepared in Section 10.3, determine the average concentration value of vinyl chloride, Cc, that corresponds to Ac, the sample peak area. Calculate the concentration of vinyl chloride in the bag, Cb, as follows:
13.0 Method Performance13.1 Analytical Range. This method is designed for the 0.1 to 50 parts per million by volume (ppmv) range. However, common gas chromatograph (GC) instruments are capable of detecting 0.02 ppmv vinyl chloride. With proper calibration, the upper limit may be extended as needed.
14.0 Pollution Prevention, [Reserved] 15.0 Waste Management, [Reserved] 16.0 References1. Brown D.W., E.W. Loy, and M.H. Stephenson. Vinyl Chloride Monitoring Near the B. F. Goodrich Chemical Company in Louisville, KY. Region IV, U.S. Environmental Protection Agency, Surveillance and Analysis Division, Athens, GA. June 24, 1974.
2. G.D. Clayton and Associates. Evaluation of a Collection and Analytical Procedure for Vinyl Chloride in Air. U.S. Environmental Protection Agency, Research Triangle Park, N.C. EPA Contract No. 68-02-1408, Task Order No. 2, EPA Report No. 75-VCL-1. December 13, 1974.
3. Midwest Research Institute. Standardization of Stationary Source Emission Method for Vinyl Chloride. U.S. Environmental Protection Agency, Research Triangle Park, N.C. Publication No. EPA-600/4-77-026. May 1977.
4. Scheil, G. and M.C. Sharp. Collaborative Testing of EPA Method 106 (Vinyl Chloride) that Will Provide for a Standardized Stationary Source Emission Measurement Method. U.S. Environmental Protection Agency, Research Triangle Park, N.C. Publication No. EPA 600/4-78-058. October 1978.
17.0 Tables, Diagrams Flowcharts, and Validation Data.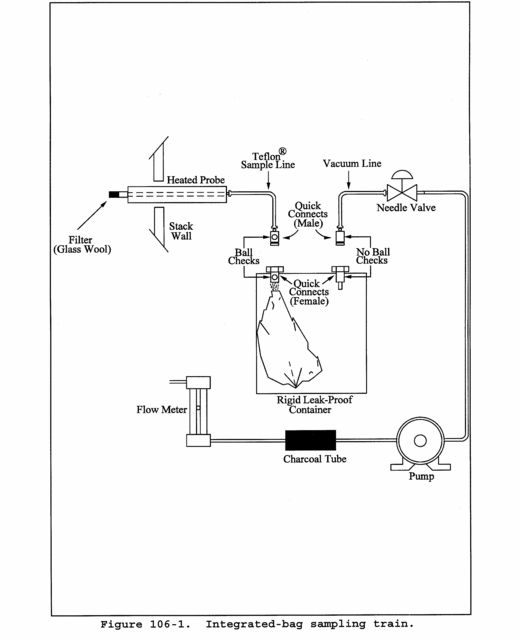 Method 107 -
Determination of Vinyl Chloride Content of In-Process Wastewater
Samples, and Vinyl Chloride Content of Polyvinyl Chloride Resin
Slurry, Wet Cake, and Latex Samples Note:
Method 107 -
Determination of Vinyl Chloride Content of In-Process Wastewater
Samples, and Vinyl Chloride Content of Polyvinyl Chloride Resin
Slurry, Wet Cake, and Latex Samples Note:
Performance of this method should not be attempted by persons unfamiliar with the operation of a gas chromatograph (GC) nor by those who are unfamiliar with source sampling, because knowledge beyond the scope of this presentation is required. This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 106.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Vinyl Chloride (CH2:CHCl) | 75-01-4 | Dependent upon analytical equipment. |
1.2 Applicability. This method is applicable for the determination of the vinyl chloride monomer (VCM) content of in-process wastewater samples, and the residual vinyl chloride monomer (RCVM) content of polyvinyl chloride (PVC) resins, wet, cake, slurry, and latex samples. It cannot be used for polymer in fused forms, such as sheet or cubes. This method is not acceptable where methods from section 304(h) of the Clean Water Act, 33 U.S.C. 1251 et seq. (the Federal Water Pollution Control Amendments of 1972 as amended by the Clean Water Act of 1977) are required.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method2.1 The basis for this method relates to the vapor equilibrium that is established at a constant known temperature in a closed system between RVCM, PVC resin, water, and air. The RVCM in a PVC resin will equilibrate rapidly in a closed vessel, provided that the temperature of the PVC resin is maintained above the glass transition temperature of that specific resin.
2.2 A sample of PVC or in-process wastewater is collected in a vial or bottle and is conditioned. The headspace in the vial or bottle is then analyzed for vinyl chloride using gas chromatography with a flame ionization detector.
3.0 Definitions [Reserved] 4.0 Interferences4.1 The chromatograph columns and the corresponding operating parameters herein described normally provide an adequate resolution of vinyl chloride; however, resolution interferences may be encountered on some sources. Therefore, the chromatograph operator shall select the column and operating parameters best suited to his particular analysis requirements, subject to the approval of the Administrator. Approval is automatic provided that confirming data are produced through an adequate supplemental analytical technique, such as analysis with a different column or GC/mass spectroscopy, and that these data are made available for review by the Administrator.
5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Toxic Analyte. Care must be exercised to prevent exposure of sampling personnel to vinyl chloride, which is a carcinogen. Do not release vinyl chloride to the laboratory atmosphere during preparation of standards. Venting or purging with VCM/air mixtures must be held to a minimum. When they are required, the vapor must be routed to outside air. Vinyl chloride, even at low ppm levels, must never be vented inside the laboratory. After vials have been analyzed, the gas must be vented prior to removal of the vial from the instrument turntable. Vials must be vented through a hypodermic needle connected to an activated charcoal tube to prevent release of vinyl chloride into the laboratory atmosphere. The charcoal must be replaced prior to vinyl chloride breakthrough.
6.0 Equipment and Supplies6.1 Sample Collection. The following equipment is required:
6.1.1 Glass bottles. 60-ml (2-oz) capacity, with wax-lined screw-on tops, for PVC samples.
6.1.2 Glass Vials. Headspace vials, with Teflon-faced butyl rubber sealing discs, for water samples.
6.1.3 Adhesive Tape. To prevent loosening of bottle tops.
6.2 Sample Recovery. The following equipment is required:
6.2.1 Glass Vials. Headspace vials, with butyl rubber septa and aluminum caps. Silicone rubber is not acceptable.
6.2.2 Analytical Balance. Capable of determining sample weight within an accuracy of ±1 percent.
6.2.3 Vial Sealer. To seal headspace vials.
6.2.4 Syringe. 100-ml capacity.
6.3 Analysis. The following equipment is required:
6.3.1 Headspace Sampler and Chromatograph. Capable of sampling and analyzing a constant amount of headspace gas from a sealed vial, while maintaining that vial at a temperature of 90 °C ±0.5 °C (194 °F ±0.9 °F). The chromatograph shall be equipped with a flame ionization detector (FID). Perkin-Elmer Corporation Models F-40, F-42, F-45, HS-6, and HS-100, and Hewlett-Packard Corporation Model 19395A have been found satisfactory. Chromatograph backflush capability may be required.
6.3.2 Chromatographic Columns. Stainless steel 1 m by 3.2 mm and 2 m by 3.2 mm, both containing 50/80-mesh Porapak Q. Other columns may be used provided that the precision and accuracy of the analysis of vinyl chloride standards are not impaired and information confirming that there is adequate resolution of the vinyl chloride peak are available for review. (Adequate resolution is defined as an area overlap of not more than 10 percent of the vinyl chloride peak by an interferant peak. Calculation of area overlap is explained in Procedure 1 of appendix C to this part: “Determination of Adequate Chromatographic Peak Resolution.”) Two 1.83 m columns, each containing 1 percent Carbowax 1500 on Carbopak B, have been found satisfactory for samples containing acetaldehyde.
6.3.3 Temperature Sensor. Range 0 to 100 °C (32 to 212 °F) accurate to 0.1 °C.
6.3.4 Integrator-Recorder. To record chromatograms.
6.3.5 Barometer. Accurate to 1 mm Hg.
6.3.6 Regulators. For required gas cylinders.
6.3.7 Headspace Vial Pre-Pressurizer. Nitrogen pressurized hypodermic needle inside protective shield.
7.0 Reagents and Standards7.1 Analysis. Same as Method 106, Section 7.1, with the addition of the following:
7.1.1 Water. Interference-free.
7.2 Calibration. The following items are required for calibration:
7.2.1 Cylinder Standards (4). Gas mixture standards (50-, 500-, 2000- and 4000-ppm vinyl chloride in nitrogen cylinders). Cylinder standards may be used directly to prepare a chromatograph calibration curve as described in Section 10.3, if the following conditions are met: (a) The manufacturer certifies the gas composition with an accuracy of ±3 percent or better (see Section 7.2.1.1). (b) The manufacturer recommends a maximum shelf life over which the gas concentration does not change by greater than ±5 percent from the certified value. (c) The manufacturer affixes the date of gas cylinder preparation, certified vinyl chloride concentration, and recommended maximum shelf life to the cylinder before shipment to the buyer.
7.2.1.1 Cylinder Standards Certification. The manufacturer shall certify the concentration of vinyl chloride in nitrogen in each cylinder by (a) directly analyzing each cylinder and (b) calibrating the analytical procedure on the day of cylinder analysis. To calibrate the analytical procedure, the manufacturer shall use, as a minimum, a 3-point calibration curve. It is recommended that the manufacturer maintain (1) a high-concentration calibration standard (between 4000 and 8000 ppm) to prepare the calibration curve by an appropriate dilution technique and (2) a low-concentration calibration standard (between 50 and 500 ppm) to verify the dilution technique used. If the difference between the apparent concentration read from the calibration curve and the true concentration assigned to the low-concentration calibration standard exceeds 5 percent of the true concentration, the manufacturer shall determine the source of error and correct it, then repeat the 3-point calibration.
7.2.1.2 Verification of Manufacturer's Calibration Standards. Before using, the manufacturer shall verify each calibration standard by (a) comparing it to gas mixtures prepared (with 99 mole percent vinyl chloride) in accordance with the procedure described in Section 10.1 of Method 106 or by (b) calibrating it against vinyl chloride cylinder Standard Reference Materials (SRMs) prepared by the National Institute of Standards and Technology, if such SRMs are available. The agreement between the initially determined concentration value and the verification concentration value must be within 5 percent. The manufacturer must reverify all calibration standards on a time interval consistent with the shelf life of the cylinder standards sold.
8.0 Sample Collection, Preservation, Storage, and Transport8.1 Sample Collection.
8.1.1 PVC Sampling. Allow the resin or slurry to flow from a tap on the tank or silo until the tap line has been well purged. Extend and fill a 60-ml sample bottle under the tap, and immediately tighten a cap on the bottle. Wrap adhesive tape around the cap and bottle to prevent the cap from loosening. Place an identifying label on each bottle, and record the date, time, and sample location both on the bottles and in a log book.
8.1.2 Water Sampling. At the sampling location fill the vials bubble-free to overflowing so that a convex meniscus forms at the top. The excess water is displaced as the sealing disc is carefully placed, with the Teflon side down, on the opening of the vial. Place the aluminum seal over the disc and the neck of the vial, and crimp into place. Affix an identifying label on the bottle, and record the date, time, and sample location both on the vials and in a log book.
8.2 Sample Storage. All samples must be analyzed within 24 hours of collection, and must be refrigerated during this period.
9.0 Quality Control| Section | Quality control measure | Effect |
|---|---|---|
| 10.3 | Chromatograph calibration | Ensure precision and accuracy of chromatograph. |
Maintain a laboratory log of all calibrations.
10.1 Preparation of Standards. Calibration standards are prepared as follows: Place 100 µl or about two equal drops of distilled water in the sample vial, then fill the vial with the VCM/nitrogen standard, rapidly seat the septum, and seal with the aluminum cap. Use a 1/8-in. stainless steel line from the cylinder to the vial. Do not use rubber or Tygon tubing. The sample line from the cylinder must be purged (into a properly vented hood) for several minutes prior to filling the vials. After purging, reduce the flow rate to between 500 and 1000 cc/min. Place end of tubing into vial (near bottom). Position a septum on top of the vial, pressing it against the 1/8-in. filling tube to minimize the size of the vent opening. This is necessary to minimize mixing air with the standard in the vial. Each vial is to be purged with standard for 90 seconds, during which time the filling tube is gradually slid to the top of the vial. After the 90 seconds, the tube is removed with the septum, simultaneously sealing the vial. Practice will be necessary to develop good technique. Rubber gloves should be worn during the above operations. The sealed vial must then be pressurized for 60 seconds using the vial prepressurizer. Test the vial for leakage by placing a drop of water on the septum at the needle hole. Prepressurization of standards is not required unless samples have been prepressurized.
10.2 Analyzer Calibration. Calibration is to be performed each 8-hour period the chromatograph is used. Alternatively, calibration with duplicate 50-, 500-, 2,000-, and 4,000-ppm standards (hereafter described as a four-point calibration) may be performed on a monthly basis, provided that a calibration confirmation test consisting of duplicate analyses of an appropriate standard is performed once per plant shift, or once per chromatograph carrousel operation (if the chromatograph operation is less frequent than once per shift). The criterion for acceptance of each calibration confirmation test is that both analyses of 500-ppm standards [2,000-ppm standards if dispersion resin (excluding latex resin) samples are being analyzed] must be within 5 percent of the most recent four-point calibration curve. If this criterion is not met, then a complete four-point calibration must be performed before sample analyses can proceed.
10.3 Preparation of Chromatograph Calibration Curve. Prepare two vials each of 50-, 500-, 2,000-, and 4,000-ppm standards. Run the calibration samples in exactly the same manner as regular samples. Plot As, the integrator area counts for each standard sample, versus Cc, the concentration of vinyl chloride in each standard sample. Draw a straight line through the points derived by the least squares method.
11.0 Analytical Procedure11.1 Preparation of Equipment. Install the chromatographic column and condition overnight at 160 °C (320 °F). In the first operation, Porapak columns must be purged for 1 hour at 230 °C (450 °F).
Do not connect the exit end of the column to the detector while conditioning. Hydrogen and air to the detector must be turned off while the column is disconnected.
11.2 Flow Rate Adjustments. Adjust flow rates as follows:
11.2.1. Nitrogen Carrier Gas. Set regulator on cylinder to read 50 psig. Set regulator on chromatograph to produce a flow rate of 30.0 cc/min. Accurately measure the flow rate at the exit end of the column using the soap film flowmeter and a stopwatch, with the oven and column at the analysis temperature. After the instrument program advances to the “B” (backflush) mode, adjust the nitrogen pressure regulator to exactly balance the nitrogen flow rate at the detector as was obtained in the “A” mode.
11.2.2. Vial Prepressurizer Nitrogen.
11.2.2.1 After the nitrogen carrier is set, solve the following equation and adjust the pressure on the vial prepressurizer accordingly.
Where: T1 = Ambient temperature, °K (°R). T2 = Conditioning bath temperature, °K (°R). P1 = Gas chromatograph absolute dosing pressure (analysis mode), k Pa. Pw1 = Water vapor pressure 525.8 mm Hg @ 90 °C. Pw2 = Water vapor pressure 19.8 mm Hg @ 22 °C. 7.50 = mm Hg per k Pa. 10 kPa = Factor to adjust the prepressurized pressure to slightly less than the dosing pressure.11.2.2.2 Because of gauge errors, the apparatus may over-pressurize the vial. If the vial pressure is at or higher than the dosing pressure, an audible double injection will occur. If the vial pressure is too low, errors will occur on resin samples because of inadequate time for head-space gas equilibrium. This condition can be avoided by running several standard gas samples at various pressures around the calculated pressure, and then selecting the highest pressure that does not produce a double injection. All samples and standards must be pressurized for 60 seconds using the vial prepressurizer. The vial is then placed into the 90 °C conditioning bath and tested for leakage by placing a drop of water on the septum at the needle hole. A clean, burr-free needle is mandatory.
11.2.3. Burner Air Supply. Set regulator on cylinder to read 50 psig. Set regulator on chromatograph to supply air to burner at a rate between 250 and 300 cc/min. Check with bubble flowmeter.
11.2.4. Hydrogen Supply. Set regulator on cylinder to read 30 psig. Set regulator on chromatograph to supply approximately 35 ±5 cc/min. Optimize hydrogen flow to yield the most sensitive detector response without extinguishing the flame. Check flow with bubble meter and record this flow.
11.3 Temperature Adjustments. Set temperatures as follows:
11.3.1. Oven (chromatograph column), 140 °C (280 °F).
11.3.2. Dosing Line, 150 °C (300 °F).
11.3.3. Injection Block, 170 °C (340 °F).
11.3.4. Sample Chamber, Water Temperature, 90 °C ±1.0 °C (194 °F ±1.8 °F).
11.4 Ignition of Flame Ionization Detector. Ignite the detector according to the manufacturer's instructions.
11.5 Amplifier Balance. Balance the amplifier according to the manufacturer's instructions.
11.6 Programming the Chromatograph. Program the chromatograph as follows:
11.6.1. I - Dosing or Injection Time. The normal setting is 2 seconds.
11.6.2. A - Analysis Time. The normal setting is approximately 70 percent of the VCM retention time. When this timer terminates, the programmer initiates backflushing of the first column.
11.6.3. B - Backflushing Time. The normal setting is double the analysis time.
11.6.4. W - Stabilization Time. The normal setting is 0.5 min to 1.0 min.
11.6.5. X - Number of Analyses Per Sample. The normal setting is one.
11.7. Sample Treatment. All samples must be recovered and analyzed within 24 hours after collection.
11.7.1 Resin Samples. The weight of the resin used must be between 0.1 and 4.5 grams. An exact weight must be obtained (within ±1 percent) for each sample. In the case of suspension resins, a volumetric cup can be prepared for holding the required amount of sample. When the cup is used, open the sample bottle, and add the cup volume of resin to the tared sample vial (tared, including septum and aluminum cap). Obtain the exact sample weight, add 100 ml or about two equal drops of water, and immediately seal the vial. Report this value on the data sheet; it is required for calculation of RVCM. In the case of dispersion resins, the cup cannot be used. Weigh the sample in an aluminum dish, transfer the sample to the tared vial, and accurately weigh it in the vial. After prepressurization of the samples, condition them for a minimum of 1 hour in the 90 °C (190 °F) bath. Do not exceed 5 hours. Prepressurization is not required if the sample weight, as analyzed, does not exceed 0.2 gram. It is also not required if solution of the prepressurization equation yields an absolute prepressurization value that is within 30 percent of the atmospheric pressure.
Note:Some aluminum vial caps have a center section that must be removed prior to placing into sample tray. If the cap is not removed, the injection needle will be damaged.
11.7.2 Suspension Resin Slurry and Wet Cake Samples. Decant the water from a wet cake sample, and turn the sample bottle upside down onto a paper towel. Wait for the water to drain, place approximately 0.2 to 4.0 grams of the wet cake sample in a tared vial (tared, including septum and aluminum cap) and seal immediately. Then determine the sample weight (1 percent). All samples weighing over 0.2 gram, must be prepressurized prior to conditioning for 1 hour at 90 °C (190 °F), except as noted in Section 11.7.1. A sample of wet cake is used to determine total solids (TS). This is required for calculating the RVCM.
11.7.3 Dispersion Resin Slurry and Latex Samples. The materials should not be filtered. Sample must be thoroughly mixed. Using a tared vial (tared, including septum and aluminum cap) add approximately eight drops (0.25 to 0.35 g) of slurry or latex using a medicine dropper. This should be done immediately after mixing. Seal the vial as soon as possible. Determine sample weight (1 percent). Condition the vial for 1 hour at 90 °C (190 °F) in the analyzer bath. Determine the TS on the slurry sample (Section 11.10).
11.7.4 In-process Wastewater Samples. Using a tared vial (tared, including septum and aluminum cap) quickly add approximately 1 cc of water using a medicine dropper. Seal the vial as soon as possible. Determine sample weight (1 percent). Condition the vial for 1 hour at 90 °C (190 °F) in the analyzer bath.
11.8 Preparation of Sample Turntable.
11.8.1 Before placing any sample into turntable, be certain that the center section of the aluminum cap has been removed. The numbered sample vials should be placed in the corresponding numbered positions in the turntable. Insert samples in the following order:
11.8.1.1 Positions 1 and 2. Old 2000-ppm standards for conditioning. These are necessary only after the analyzer has not been used for 24 hours or longer.
11.8.1.2 Position 3. 50-ppm standard, freshly prepared.
11.8.1.3 Position 4. 500-ppm standard, freshly prepared.
11.8.1.4 Position 5. 2000-ppm standard, freshly prepared.
11.8.1.5 Position 6. 4000-ppm standard, freshly prepared.
11.8.1.6 Position 7. Sample No. 7 (This is the first sample of the day, but is given as 7 to be consistent with the turntable and the integrator printout.)
11.8.2 After all samples have been positioned, insert the second set of 50-, 500-, 2000-, and 4000-ppm standards. Samples, including standards, must be conditioned in the bath of 90 °C (190 °F) for a minimum of one hour and a maximum of five hours.
11.9 Start Chromatograph Program. When all samples, including standards, have been conditioned at 90 °C (190 °F) for at least one hour, start the analysis program according to the manufacturer's instructions. These instructions must be carefully followed when starting and stopping a program to prevent damage to the dosing assembly.
11.10 Determination of Total Solids. For wet cake, slurry, resin solution, and PVC latex samples, determine TS for each sample by accurately weighing approximately 3 to 4 grams of sample in an aluminum pan before and after placing in a draft oven (105 to 110 °C (221 to 230 °F)). Samples must be dried to constant weight. After first weighing, return the pan to the oven for a short period of time, and then reweigh to verify complete dryness. The TS are then calculated as the final sample weight divided by initial sample weight.
12.0 Calculations and Data Analysis12.1 Nomenclature.
As = Chromatogram area counts of vinyl chloride for the sample, area counts. As = Chromatogram area counts of vinyl chloride for the sample. Cc = Concentration of vinyl chloride in the standard sample, ppm. Kp = Henry's Law Constant for VCM in PVC 90 °C, 6.52 × 10−6 g/g/mm Hg. Kw = Henry's Law Constant for VCM in water 90 °C, 7 × 10−7 g/g/mm Hg. Mv = Molecular weight of VCM, 62.5 g/mole. m = Sample weight, g. Pa = Ambient atmospheric pressure, mm Hg. R = Gas constant, (62360 3 ml) (mm Hg)/(mole)(°K). Rf = Response factor in area counts per ppm VCM. Rs = Response factor, area counts/ppm. Tl = Ambient laboratory temperature, °K. TS = Total solids expressed as a decimal fraction. T2 = Equilibrium temperature, °K. Vg = Volume of vapor phase, ml. Vv = Vial volume, 3 ml. 1.36 = Density of PVC at 90 °C, g/ 3 ml. 0.9653 = Density of water at 90 °C, g/ 3 ml.12.2 Response Factor. If the calibration curve described in Section 10.3 passes through zero, an average response factor, Rf, may be used to facilitate computation of vinyl chloride sample concentrations.
12.2.1 To compute Rf, first compute a response factor, Rs, for each sample as follows:
12.2.2 Sum the individual response factors, and calculate Rf. If the calibration curve does not pass through zero, use the calibration curve to determine each sample concentration.
12.3 Residual Vinyl Chloride Monomer Concentration, (Crvc) or Vinyl Chloride Monomer Concentration. Calculate Crvc in ppm or mg/kg as follows:
 Note:
Note:
Results calculated using these equations represent concentration based on the total sample. To obtain results based on dry PVC content, divide by TS.
13.0 Method Performance13.1 Range and Sensitivity. The lower limit of detection of vinyl chloride will vary according to the sampling and chromatographic system. The system should be capable of producing a measurement for a 50-ppm vinyl chloride standard that is at least 10 times the standard deviation of the system background noise level.
13.2 An interlaboratory comparison between seven laboratories of three resin samples, each split into three parts, yielded a standard deviation of 2.63 percent for a sample with a mean of 2.09 ppm, 4.16 percent for a sample with a mean of 1.66 ppm, and 5.29 percent for a sample with a mean of 62.66 ppm.
14.0 Pollution Prevention [Reserved] 15.0 Waste Management [Reserved] 16.0 References1. B.F. Goodrich, Residual Vinyl Chloride Monomer Content of Polyvinyl Chloride Resins, Latex, Wet Cake, Slurry and Water Samples. B.F. Goodrich Chemical Group Standard Test Procedure No. 1005-E. B.F. Goodrich Technical Center, Avon Lake, Ohio. October 8, 1979.
2. Berens, A.R. The Diffusion of Vinyl Chloride in Polyvinyl Chloride. ACS-Division of Polymer Chemistry, Polymer Preprints 15 (2):197. 1974.
3. Berens, A.R. The Diffusion of Vinyl Chloride in Polyvinyl Chloride. ACS-Division of Polymer Chemistry, Polymer Preprints 15 (2):203. 1974.
4. Berens, A.R., et. al. Analysis for Vinyl Chloride in PVC Powders by Head-Space Gas Chromatography. Journal of Applied Polymer Science. 19:3169-3172. 1975.
5. Mansfield, R.A. The Evaluation of Henry's Law Constant (Kp) and Water Enhancement in the Perkin-Elmer Multifract F-40 Gas Chromatograph. B.F. Goodrich. Avon Lake, Ohio. February 10, 1978.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved] Method 107A - Determination of Vinyl Chloride Content of Solvents, Resin-Solvent Solution, Polyvinyl Chloride Resin, Resin Slurry, Wet Resin, and Latex Samples IntroductionPerformance of this method should not be attempted by persons unfamiliar with the operation of a gas chromatograph (GC) or by those who are unfamiliar with source sampling because knowledge beyond the scope of this presentation is required. Care must be exercised to prevent exposure of sampling personnel to vinyl chloride, a carcinogen.
1. Applicability and Principle1.1 Applicability. This is an alternative method and applies to the measurement of the vinyl chloride content of solvents, resin solvent solutions, polyvinyl chloride (PVC) resin, wet cake slurries, latex, and fabricated resin samples. This method is not acceptable where methods from Section 304(h) of the Clean Water Act, 33 U.S.C. 1251et seq., (the Federal Water Pollution Control Act Amendments of 1972 as amended by the Clean Water Act of 1977) are required.
1.2 Principle. The basis for this method lies in the direct injection of a liquid sample into a chromatograph and the subsequent evaporation of all volatile material into the carrier gas stream of the chromatograph, thus permitting analysis of all volatile material including vinyl chloride.
2. Range and SensitivityThe lower limit of detection of vinyl chloride in dry PVC resin is 0.2 ppm. For resin solutions, latexes, and wet resin, this limit rises inversely as the nonvolatile (resin) content decreases.
With proper calibration, the upper limit may be extended as needed.
3. InterferencesThe chromatograph columns and the corresponding operating parameters herein described normally provide an adequate resolution of vinyl chloride. In cases where resolution interferences are encountered, the chromatograph operator shall select the column and operating parameters best suited to his particular analysis problem, subject to the approval of the Administrator. Approval is automatic, provided that the tester produces confirming data through an adequate supplemental analytical technique, such as analysis with a different column or GC/mass spectroscopy, and has the data available for review by the Administrator.
4. Precision and ReproducibilityA standard sample of latex containing 181.8 ppm vinyl chloride analyzed 10 times by the alternative method showed a standard deviation of 7.5 percent and a mean error of 0.21 percent.
A sample of vinyl chloride copolymer resin solution was analyzed 10 times by the alternative method and showed a standard deviation of 6.6 percent at a level of 35 ppm.
5. SafetyDo not release vinyl chloride to the laboratory atmosphere during preparation of standards. Venting or purging with vinyl chloride monomer (VCM) air mixtures must be held to minimum. When purging is required, the vapor must be routed to outside air. Vinyl chloride, even at low-ppm levels, must never be vented inside the laboratory.
6. Apparatus6.1 Sampling. The following equipment is required:
6.1.1 Glass Bottles. 16-oz wide mouth wide polyethylene-lined, screw-on tops.
6.1.2 Adhesive Tape. To prevent loosening of bottle tops.
6.2 Sample Recovery. The following equipment is required:
6.2.1 Glass Vials. 20-ml capacity with polycone screw caps.
6.2.2 Analytical Balance. Capable of weighing to ±0.01 gram.
6.2.3 Syringe. 50-microliter size, with removable needle.
6.2.4 Fritted Glass Sparger. Fine porosity.
6.2.5 Aluminum Weighing Dishes.
6.2.6 Sample Roller or Shaker. To help dissolve sample.
6.3 Analysis. The following equipment is required:
6.3.1 Gas Chromatograph. Hewlett Packard Model 5720A or equivalent.
6.3.2 Chromatograph Column. Stainless steel, 6.1 m by 3.2 mm, packed with 20 percent Tergitol E-35 on Chromosorb W AW 60/80 mesh. The analyst may use other columns provided that the precision and accuracy of the analysis of vinyl chloride standards are not impaired and that he has available for review information confirming that there is adequate resolution of the vinyl chloride peak. (Adequate resolution is defined as an area overlap of not more than 10 percent of the vinyl chloride peak by an interfering peak. Calculation of area overlap is explained in Appendix C, Procedure 1: “Determination of Adequate Chromatographic Peak Resolution.”)
6.3.3 Valco Instrument Six-Port Rotary Valve. For column back flush.
6.3.4 Septa. For chromatograph injection port.
6.3.5 Injection Port Liners. For chromatograph used.
6.3.6 Regulators. For required gas cylinders.
6.3.7 Soap Film Flowmeter. Hewlett Packard No. 0101-0113 or equivalent.
6.4 Calibration. The following equipment is required:
6.4.1 Analytical Balance. Capable of weighing to ±0.0001 g.
6.4.2 Erlenmeyer Flask With Glass Stopper. 125 ml.
6.4.3 Pipets. 0.1, 0.5, 1, 5, 10, and 50 ml.
6.4.4 Volumetric Flasks. 10 and 100 ml.
7. ReagentsUse only reagents that are of chromatograph grade.
7.1 Analysis. The following items are required:
7.1.1 Hydrogen Gas. Zero grade.
7.1.2 Nitrogen Gas. Zero grade.
7.1.3 Air. Zero grade.
7.1.4 Tetrahydrofuran (THF). Reagent grade.
Analyze the THF by injecting 10 microliters into the prepared gas chromatograph. Compare the THF chromatogram with that shown in Figure 107A-1. If the chromatogram is comparable to A, the THF should be sparged with pure nitrogen for approximately 2 hours using the fritted glass sparger to attempt to remove the interfering peak. Reanalyze the sparged THF to determine whether the THF is acceptable for use. If the scan is comparable to B, the THF should be acceptable for use in the analysis.
7.1.5 N, N-Dimethylacetamide (DMAC). Spectrographic grade. For use in place of THF.
7.2 Calibration. The following item is required:
7.2.1 Vinyl Chloride 99.9 Percent. Ideal Gas Products lecture bottle, or equivalent. For preparation of standard solutions.
8. Procedure8.1 Sampling. Allow the liquid or dried resin to flow from a tap on the tank, silo, or pipeline until the tap has been purged. Fill a wide-mouth pint bottle, and immediately tightly cap the bottle. Place an identifying label on each bottle and record the date, time, sample location, and material.
8.2 Sample Treatment. Sample must be run within 24 hours.
8.2.1 Resin Samples. Weigh 9.00 ±0.01 g of THF or DMAC in a tared 20-ml vial. Add 1.00 ±0.01 g of resin to the tared vial containing the THF or DMAC. Close the vial tightly with the screw cap, and shake or otherwise agitate the vial until complete solution of the resin is obtained. Shaking may require several minutes to several hours, depending on the nature of the resin.
8.2.2 Suspension Resin Slurry and Wet Resin Sample. Slurry must be filtered using a small Buchner funnel with vacuum to yield a wet resin sample. The filtering process must be continued only as long as a steady stream of water is exiting from the funnel. Excessive filtration time could result in some loss of VCM. The wet resin sample is weighed into a tared 20-ml vial with THF or DMAC as described earlier for resin samples (8.2.1) and treated the same as the resin sample. A sample of the wet resin is used to determine total solids as required for calculating the residual VCM (Section 8.3.4).
8.2.3 Latex and Resin Solvent Solutions. Samples must be thoroughly mixed. Weigh 1.00 ±0.01 g of the latex or resin-solvent solution into a 20-ml vial containing 9.00 ±0.01 g of THF or DMAC as for the resin samples (8.2.1). Cap and shake until complete solution is obtained. Determine the total solids of the latex or resin solution sample (Section 8.3.4).
8.2.4 Solvents and Non-viscous Liquid Samples. No preparation of these samples is required. The neat samples are injected directly into the GC.
8.3 Analysis.
8.3.1 Preparation of GC. Install the chromatographic column, and condition overnight at 70 °C. Do not connect the exit end of the column to the detector while conditioning.
8.3.1.1 Flow Rate Adjustments. Adjust the flow rate as follows:
a. Nitrogen Carrier Gas. Set regulator on cylinder to read 60 psig. Set column flow controller on the chromatograph using the soap film flowmeter to yield a flow rate of 40 cc/min.
b. Burner Air Supply. Set regulator on the cylinder at 40 psig. Set regulator on the chromatograph to supply air to the burner to yield a flow rate of 250 to 300 cc/min using the flowmeter.
c. Hydrogen. Set regulator on cylinder to read 60 psig. Set regulator on the chromatograph to supply 30 to 40 cc/min using the flowmeter. Optimize hydrogen flow to yield the most sensitive detector response without extinguishing the flame. Check flow with flowmeter and record this flow.
d. Nitrogen Back Flush Gas. Set regulator on the chromatograph using the soap film flowmeter to yield a flow rate of 40 cc/min.
8.3.1.2 Temperature Adjustments. Set temperature as follows:
a. Oven (chromatographic column) at 70 °C.
b. Injection Port at 100 °C.
c. Detector at 300 °C.
8.3.1.3 Ignition of Flame Ionization Detector. Ignite the detector according to the manufacturer's instructions. Allow system to stabilize approximately 1 hour.
8.3.1.4 Recorder. Set pen at zero and start chart drive.
8.3.1.5 Attenuation. Set attenuation to yield desired peak height depending on sample VCM content.
8.3.2 Chromatographic Analyses.
a. Sample Injection. Remove needle from 50-microliter syringe. Open sample vial and draw 50-microliters of THF or DMAC sample recovery solution into the syringe. Recap sample vial. Attach needle to the syringe and while holding the syringe vertically (needle uppermost), eject 40 microliters into an absorbent tissue. Wipe needle with tissue. Now inject 10 microliters into chromatograph system. Repeat the injection until two consecutive values for the height of the vinyl chloride peak do not vary more than 5 percent. Use the average value for these two peak heights to compute the sample concentration.
b. Back Flush. After 4 minutes has elapsed after sample injection, actuate the back flush valve to purge the first 4 feet of the chromatographic column of solvent and other high boilers.
c. Sample Data. Record on the chromatograph strip chart the data from the sample label.
d. Elution Time. Vinyl chloride elutes at 2.8 minutes. Acetaldehyde elutes at 3.7 minutes. Analysis is considered complete when chart pen becomes stable. After 5 minutes, reset back flush valve and inject next sample.
8.3.3 Chromatograph Servicing.
a. Septum. Replace after five sample injections.
b. Sample Port Liner. Replace the sample port liner with a clean spare after five sample injections.
c. Chromatograph Shutdown. If the chromatograph has been shut down overnight, rerun one or more samples from the preceding day to test stability and precision prior to starting on the current day's work.
8.3.4 Determination of Total Solids (TS). For wet resin, resin solution, and PVC latex samples, determine the TS for each sample by accurately weighing approximately 3 to 5 grams of sample into a tared aluminum pan. The initial procedure is as follows:
a. Where water is the major volatile component: Tare the weighing dish, and add 3 to 5 grams of sample to the dish. Weigh to the nearest milligram.
b. Where volatile solvent is the major volatile component: Transfer a portion of the sample to a 20-ml screw cap vial and cap immediately. Weigh the vial to the nearest milligram. Uncap the vial and transfer a 3- to 5-gram portion of the sample to a tared aluminum weighing dish. Recap the vial and reweigh to the nearest milligram. The vial weight loss is the sample weight.
To continue, place the weighing pan in a 130 °C oven for 1 hour. Remove the dish and allow to cool to room temperature in a desiccator. Weigh the pan to the nearest 0.1 mg. Total solids is the weight of material in the aluminum pan after heating divided by the net weight of sample added to the pan originally times 100.
9. Calibration of the Chromatograph9.1 Preparation of Standards. Prepare a 1 percent by weight (approximate) solution of vinyl chloride in THF or DMAC by bubbling vinyl chloride gas from a cylinder into a tared 125-ml glass-stoppered flask containing THF or DMAC. The weight of vinyl chloride to be added should be calculated prior to this operation, i.e., 1 percent of the weight of THF or DMAC contained in the tared flask. This must be carried out in a laboratory hood. Adjust the vinyl chloride flow from the cylinder so that the vinyl chloride dissolves essentially completely in the THF or DMAC and is not blown to the atmosphere. Take particular care not to volatize any of the solution. Stopper the flask and swirl the solution to effect complete mixing. Weigh the stoppered flask to nearest 0.1 mg to determine the exact amount of vinyl chloride added.
Pipet 10 ml of the approximately 1 percent solution into a 100-ml glass-stoppered volumetric flask, and add THF or DMAC to fill to the mark. Cap the flask and invert 10 to 20 times. This solution contains approximately 1,000 ppm by weight of vinyl chloride (note the exact concentration).
Pipet 50-, 10-, 5-, 1-, 0.5-, and 0.1-ml aliquots of the approximately 1,000 ppm solution into 10 ml glass stoppered volumetric flasks. Dilute to the mark with THF or DMAC, cap the flasks and invert each 10 to 20 times. These solutions contain approximately 500, 100, 50, 10, 5, and 1 ppm vinyl chloride. Note the exact concentration of each one. These standards are to be kept under refrigeration in stoppered bottles, and must be renewed every 3 months.
9.2 Preparation of Chromatograph Calibration Curve.
Obtain the GC for each of the six final solutions prepared in Section 9.1 by using the procedure in Section 8.3.2. Prepare a chart plotting peak height obtained from the chromatogram of each solution versus the known concentration. Draw a straight line through the points derived by the least squares method.
10. Calculations10.1 Response Factor. From the calibration curve described in Section 9.2, select the value of Cc that corresponds to Hc for each sample. Compute the response factor, Rf, for each sample as follows:
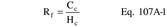 where: Rf
= Chromatograph response factor, ppm/mm. Cc = Concentration of
vinyl chloride in the standard sample, ppm. Hc = Peak height of the
standard sample, mm.
where: Rf
= Chromatograph response factor, ppm/mm. Cc = Concentration of
vinyl chloride in the standard sample, ppm. Hc = Peak height of the
standard sample, mm.
10.2 Residual vinyl chloride monomer concentration (Crvc) or vinyl chloride monomer concentration in resin:
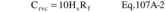 Where:
Crvc = Concentration of residual vinyl chloride monomer, ppm. Hs =
Peak height of sample, mm. Rf = Chromatograph response factor.
Where:
Crvc = Concentration of residual vinyl chloride monomer, ppm. Hs =
Peak height of sample, mm. Rf = Chromatograph response factor.
10.3 Samples containing volatile material, i.e., resin solutions, wet resin, and latexes:
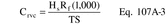 where: TS
= Total solids in the sample, weight fraction.
where: TS
= Total solids in the sample, weight fraction.
10.4 Samples of solvents and in process wastewater:
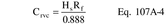 Where:
0.888 = Specific gravity of THF. 11. Bibliography
Where:
0.888 = Specific gravity of THF. 11. Bibliography
1. Communication from R. N. Wheeler, Jr.; Union Carbide Corporation. Part 61 National Emissions Standards for Hazardous Air Pollutants appendix B, Method 107 - Alternate Method, September 19, 1977.
Method 108 - Determination of Particulate and Gaseous Arsenic Emissions Note:This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in appendix A to 40 CFR part 60. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, Method 5, and Method 12.
1.0 Scope and Application.1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Arsenic compounds as arsenic (As) | 7440-38-2 | Lower limit 10 µg/ml or less. |
1.2 Applicability. This method is applicable for the determination of inorganic As emissions from stationary sources as specified in an applicable subpart of the regulations.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of MethodParticulate and gaseous As emissions are withdrawn isokinetically from the source and are collected on a glass mat filter and in water. The collected arsenic is then analyzed by means of atomic absorption spectrophotometry (AAS).
3.0 Definitions. [Reserved] 4.0 InterferencesAnalysis for As by flame AAS is sensitive to the chemical composition and to the physical properties (e.g., viscosity, pH) of the sample. The analytical procedure includes a check for matrix effects (Section 11.5).
5.0 Safety5.1 This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive reagents. The following reagents are hazardous. Personal protective equipment and safe procedures that prevent chemical splashes are recommended. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrochloric Acid (HCl). Highly corrosive liquid with toxic vapors. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Hydrogen Peroxide (H2O2). Very harmful to eyes. 30% H2O2 can burn skin, nose, and lungs.
5.2.3 Nitric Acid (HNO3). Highly corrosive to eyes, skin, nose, and lungs. Vapors are highly toxic and can cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.4 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with small amounts of water.
6.0 Equipment and Supplies6.1 Sample Collection. A schematic of the sampling train used in performing this method is shown in Figure 108-1; it is similar to the Method 5 sampling train of 40 CFR part 60, appendix A. The following items are required for sample collection:
6.1.1 Probe Nozzle, Probe Liner, Pitot Tube, Differential Pressure Gauge, Filter Holder, Filter Heating System, Temperature Sensor, Metering System, Barometer, and Gas Density Determination Equipment. Same as Method 5, Sections 6.1.1.1 to 6.1.1.7, 6.1.1.9, 6.1.2, and 6.1.3, respectively.
6.1.2 Impingers. Four impingers connected in series with leak-free ground-glass fittings or any similar leak-free noncontaminating fittings. For the first, third, and fourth impingers, use the Greenburg-Smith design, modified by replacing the tip with a 1.3-cm ID (0.5-in.) glass tube extending to about 1.3 cm (0.5 in.) from the bottom of the flask. For the second impinger, use the Greenburg-Smith design with the standard tip. Modifications (e.g., flexible connections between the impingers, materials other than glass, or flexible vacuum lines to connect the filter holder to the condenser) are subject to the approval of the Administrator.
6.1.3 Temperature Sensor. Place a temperature sensor, capable of measuring temperature to within 1 °C (2 °F), at the outlet of the fourth impinger for monitoring purposes.
6.2 Sample Recovery. The following items are required for sample recovery:
6.2.1 Probe-Liner and Probe-Nozzle Brushes, Petri Dishes, Graduated Cylinder and/or Balance, Plastic Storage Containers, and Funnel and Rubber Policeman. Same as Method 5, Sections 6.2.1 and 6.2.4 to 6.2.8, respectively.
6.2.2 Wash Bottles. Polyethylene (2).
6.2.3 Sample Storage Containers. Chemically resistant, polyethylene or polypropylene for glassware washes, 500- or 1000-ml.
6.3 Analysis. The following items are required for analysis:
6.3.1 Spectrophotometer. Equipped with an electrodeless discharge lamp and a background corrector to measure absorbance at 193.7 nanometers (nm). For measuring samples having less than 10 µg As/ml, use a vapor generator accessory or a graphite furnace.
6.3.2 Recorder. To match the output of the spectrophotometer.
6.3.3 Beakers. 150 ml.
6.3.4 Volumetric Flasks. Glass 50-, 100-, 200-, 500-, and 1000-ml; and polypropylene, 50-ml.
6.3.5 Balance. To measure within 0.5 g.
6.3.6 Volumetric Pipets. 1-, 2-, 3-, 5-, 8-, and 10-ml.
6.3.7 Oven.
6.3.8 Hot Plate.
7.0 Reagents and StandardsUnless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 The following reagents are required for sample collection:
7.1.1 Filters. Same as Method 5, Section 7.1.1, except that the filters need not be unreactive to SO2.
7.1.2 Silica Gel, Crushed Ice, and Stopcock Grease. Same as Method 5, Sections 7.1.2, 7.1.4, and 7.1.5, respectively.
7.1.3 Water. Deionized distilled to meet ASTM D 1193-77 or 91 (incorporated by reference-see § 61.18), Type 3. When high concentrations of organic matter are not expected to be present, the KMnO4 test for oxidizable organic matter may be omitted.
7.2 Sample Recovery.
7.2.1 0.1 N NaOH. Dissolve 4.00 g of NaOH in about 500 ml of water in a 1-liter volumetric flask. Then, dilute to exactly 1.0 liter with water.
7.3 Analysis. The following reagents and standards are required for analysis:
7.3.1 Water. Same as Section 7.1.3.
7.3.2 Sodium Hydroxide, 0.1 N. Same as in Section 7.2.1.
7.3.3 Sodium Borohydride (NaBH4), 5 Percent Weight by Volume (W/V). Dissolve 50.0 g of NaBH4 in about 500 ml of 0.1 N NaOH in a 1-liter volumetric flask. Then, dilute to exactly 1.0 liter with 0.1 N NaOH.
7.3.4 Hydrochloric Acid, Concentrated.
7.3.5 Potassium Iodide (KI), 30 Percent (W/V). Dissolve 300 g of KI in 500 ml of water in a 1 liter volumetric flask. Then, dilute to exactly 1.0 liter with water.
7.3.6 Nitric Acid, Concentrated.
7.3.7 Nitric Acid, 0.8 N. Dilute 52 ml of concentrated HNO3 to exactly 1.0 liter with water.
7.3.8 Nitric Acid, 50 Percent by Volume (V/V). Add 50 ml concentrated HNO3 to 50 ml water.
7.3.9 Stock Arsenic Standard, 1 mg As/ml. Dissolve 1.3203 g of primary standard grade As2O3 in 20 ml of 0.1 N NaOH in a 150 ml beaker. Slowly add 30 ml of concentrated HNO3. Heat the resulting solution and evaporate just to dryness. Transfer the residue quantitatively to a 1-liter volumetric flask, and dilute to 1.0 liter with water.
7.3.10 Arsenic Working Solution, 1.0 µg As/ml. Pipet exactly 1.0 ml of stock arsenic standard into an acid-cleaned, appropriately labeled 1-liter volumetric flask containing about 500 ml of water and 5 ml of concentrated HNO3. Dilute to exactly 1.0 liter with water.
7.3.11 Air. Suitable quality for AAS analysis.
7.3.12 Acetylene. Suitable quality for AAS analysis.
7.3.13 Nickel Nitrate, 5 Percent Ni (W/V). Dissolve 24.780 g of nickel nitrate hexahydrate [Ni(NO3)26H2O] in water in a 100-ml volumetric flask, and dilute to 100 ml with water.
7.3.14 Nickel Nitrate, 1 Percent Ni (W/V). Pipet 20 ml of 5 percent nickel nitrate solution into a 100-ml volumetric flask, and dilute to exactly 100 ml with water.
7.3.15 Hydrogen Peroxide, 3 Percent by Volume. Pipet 50 ml of 30 percent H2O2 into a 500-ml volumetric flask, and dilute to exactly 500 ml with water.
8.0 Sample Collection, Preservation, Transport, and Storage8.1 Pretest Preparation. Follow the general procedure given in Method 5, Section 8.1, except the filter need not be weighed, and the 200 ml of 0.1N NaOH and Container 4 should be tared to within 0.5 g.
8.2 Preliminary Determinations. Follow the general procedure given in Method 5, Section 8.2, except select the nozzle size to maintain isokinetic sampling rates below 28 liters/min (1.0 cfm).
8.3 Preparation of Sampling Train. Follow the general procedure given in Method 5, Section 8.3.
8.4 Leak-Check Procedures. Same as Method 5, Section 8.4.
8.5 Sampling Train Operation. Follow the general procedure given in Method 5, Section 8.5, except maintain isokinetic sampling flow rates below 28 liters/min (1.0 cfm). For each run, record the data required on a data sheet similar to the one shown in Figure 108-2.
8.6 Calculation of Percent Isokinetic. Same as Method 5, Section 8.6.
8.7 Sample Recovery. Same as Method 5, Section 8.7, except that 0.1 N NaOH is used as the cleanup solvent instead of acetone and that the impinger water is treated as follows:
8.7.1 Container Number 4 (Impinger Water). Clean each of the first three impingers and connecting glassware in the following manner:
8.7.1.1 Wipe the impinger ball joints free of silicone grease, and cap the joints.
8.7.1.2 Rotate and agitate each of the first two impingers, using the impinger contents as a rinse solution.
8.7.1.3 Transfer the liquid from the first three impingers to Container Number 4. Remove the outlet ball-joint cap, and drain the contents through this opening. Do not separate the impinger parts (inner and outer tubes) while transferring their contents to the container.
8.7.1.4 Weigh the contents of Container No. 4 to within 0.5 g. Record in the log the weight of liquid along with a notation of any color or film observed in the impinger catch. The weight of liquid is needed along with the silica gel data to calculate the stack gas moisture content.
Note:Measure and record the total amount of 0.1 N NaOH used for rinsing under Sections 8.7.1.5 and 8.7.1.6.
8.7.1.5 Pour approximately 30 ml of 0.1 NaOH into each of the first two impingers, and agitate the impingers. Drain the 0.1 N NaOH through the outlet arm of each impinger into Container Number 4. Repeat this operation a second time; inspect the impingers for any abnormal conditions.
8.7.1.6 Wipe the ball joints of the glassware connecting the impingers and the back half of the filter holder free of silicone grease, and rinse each piece of glassware twice with 0.1 N NaOH; transfer this rinse into Container Number 4. (DO NOT RINSE or brush the glass-fritted filter support.) Mark the height of the fluid level to determine whether leakage occurs during transport. Label the container to identify clearly its contents.
8.8 Blanks.
8.8.1 Sodium Hydroxide. Save a portion of the 0.1 N NaOH used for cleanup as a blank. Take 200 ml of this solution directly from the wash bottle being used and place it in a plastic sample container labeled “NaOH blank.”
8.8.2 Water. Save a sample of the water, and place it in a container labeled “H2O blank.”
8.8.3 Filter. Save two filters from each lot of filters used in sampling. Place these filters in a container labeled “filter blank.”
9.0 Quality Control 9.1 Miscellaneous Quality Control Measures.| Section | Quality control measure | Effect |
|---|---|---|
| 8.4, 10.1 | Sampling equipment leak-checks and calibration | Ensures accuracy and precision of sampling measurements. |
| 10.4 | Spectrophotometer calibration | Ensures linearity of spectrophotometer response to standards. |
| 11.5 | Check for matrix effects | Eliminates matrix effects. |
9.2 Volume Metering System Checks. Same as Method 5, Section 9.2.
10.0 Calibration and Standardization Note:Maintain a laboratory log of all calibrations.
10.1 Sampling Equipment. Same as Method 5, Section 10.0.
10.2 Preparation of Standard Solutions.
10.2.1 For the high level procedure, pipet 1, 3, 5, 8, and 10 ml of the 1.0 mg As/ml stock solution into separate 100 ml volumetric flasks, each containing 5 ml of concentrated HNO3. Dilute to the mark with water.
10.2.2 For the low level vapor generator procedure, pipet 1, 2, 3, and 5 ml of 1.0 µg As/ml standard solution into separate reaction tubes. Dilute to the mark with water.
10.2.3 For the low level graphite furnace procedure, pipet 1, 5, 10 and 15 ml of 1.0 µg As/ml standard solution into separate flasks along with 2 ml of the 5 percent nickel nitrate solution and 10 ml of the 3 percent H2O2 solution. Dilute to the mark with water.
10.3 Calibration Curve. Analyze a 0.8 N HNO3 blank and each standard solution according to the procedures outlined in section 11.4.1. Repeat this procedure on each standard solution until two consecutive peaks agree within 3 percent of their average value. Subtract the average peak height (or peak area) of the blank - which must be less than 2 percent of recorder full scale - from the averaged peak height of each standard solution. If the blank absorbance is greater than 2 percent of full-scale, the probable cause is As contamination of a reagent or carry-over of As from a previous sample. Prepare the calibration curve by plotting the corrected peak height of each standard solution versus the corresponding final total As weight in the solution.
10.4 Spectrophotometer Calibration Quality Control. Calculate the least squares slope of the calibration curve. The line must pass through the origin or through a point no further from the origin than ±2 percent of the recorder full scale. Multiply the corrected peak height by the reciprocal of the least squares slope to determine the distance each calibration point lies from the theoretical calibration line. The difference between the calculated concentration values and the actual concentrations (e.g., 1, 3, 5, 8, and 10 mg As for the high-level procedure) must be less than 7 percent for all standards.
Note:For instruments equipped with direct concentration readout devices, preparation of a standard curve will not be necessary. In all cases, follow calibration and operational procedures in the manufacturers' instruction manual.
11.0 Analytical Procedure11.1 Sample Loss Check. Prior to analysis, check the liquid level in Containers Number 2 and Number 4. Note on the analytical data sheet whether leakage occurred during transport. If a noticeable amount of leakage occurred, either void the sample or take steps, subject to the approval of the Administrator, to adjust the final results.
11.2 Sample Preparation.
11.2.1 Container Number 1 (Filter). Place the filter and loose particulate matter in a 150 ml beaker. Also, add the filtered solid material from Container Number 2 (see Section 11.2.2). Add 50 ml of 0.1 N NaOH. Then stir and warm on a hot plate at low heat (do not boil) for about 15 minutes. Add 10 ml of concentrated HNO3, bring to a boil, then simmer for about 15 minutes. Filter the solution through a glass fiber filter. Wash with hot water, and catch the filtrate in a clean 150 ml beaker. Boil the filtrate, and evaporate to dryness. Cool, add 5 ml of 50 percent HNO3, and then warm and stir. Allow to cool. Transfer to a 50-ml volumetric flask, dilute to volume with water, and mix well.
11.2.2 Container Number 2 (Probe Wash).
11.2.2.1 Filter (using a glass fiber filter) the contents of Container Number 2 into a 200 ml volumetric flask. Combine the filtered (solid) material with the contents of Container Number 1 (Filter).
11.2.2.2 Dilute the filtrate to exactly 200 ml with water. Then pipet 50 ml into a 150 ml beaker. Add 10 ml of concentrated HNO3, bring to a boil, and evaporate to dryness. Allow to cool, add 5 ml of 50 percent HNO3, and then warm and stir. Allow the solution to cool, transfer to a 50-ml volumetric flask, dilute to volume with water, and mix well.
11.2.3 Container Number 4 (Impinger Solution). Transfer the contents of Container Number 4 to a 500 ml volumetric flask, and dilute to exactly 500-ml with water. Pipet 50 ml of the solution into a 150-ml beaker. Add 10 ml of concentrated HNO3, bring to a boil, and evaporate to dryness. Allow to cool, add 5 ml of 50 percent HNO3, and then warm and stir. Allow the solution to cool, transfer to a 50-ml volumetric flask, dilute to volume with water, and mix well.
11.2.4 Filter Blank. Cut each filter into strips, and treat each filter individually as directed in Section 11.2.1, beginning with the sentence, “Add 50 ml of 0.1 N NaOH.”
11.2.5 Sodium Hydroxide and Water Blanks. Treat separately 50 ml of 0.1 N NaOH and 50 ml water, as directed under Section 11.2.3, beginning with the sentence, “Pipet 50 ml of the solution into a 150-ml beaker.”
11.3 Spectrophotometer Preparation. Turn on the power; set the wavelength, slit width, and lamp current. Adjust the background corrector as instructed by the manufacturer's manual for the particular atomic absorption spectrophotometer. Adjust the burner and flame characteristics as necessary.
11.4 Analysis. Calibrate the analytical equipment and develop a calibration curve as outlined in Sections 10.2 through 10.4.
11.4.1 Arsenic Samples. Analyze an appropriately sized aliquot of each diluted sample (from Sections 11.2.1 through 11.2.3) until two consecutive peak heights agree within 3 percent of their average value. If applicable, follow the procedures outlined in Section 11.4.1.1. If the sample concentration falls outside the range of the calibration curve, make an appropriate dilution with 0.8 N HNO3 so that the final concentration falls within the range of the curve. Using the calibration curve, determine the arsenic concentration in each sample fraction.
Note:Because instruments vary between manufacturers, no detailed operating instructions will be given here. Instead, the instrument manufacturer's detailed operating instructions should be followed.
11.4.1.1 Arsenic Determination at Low Concentration. The lower limit of flame AAS is 10 µg As/ml. If the arsenic concentration of any sample is at a lower level, use the graphite furnace or vapor generator which is available as an accessory component. Flame, graphite furnace, or vapor generators may be used for samples whose concentrations are between 10 and 30 µg/ml. Follow the manufacturer's instructions in the use of such equipment.
11.4.1.1.1 Vapor Generator Procedure. Place a sample containing between 0 and 5 µg of arsenic in the reaction tube, and dilute to 15 ml with water. Since there is some trial and error involved in this procedure, it may be necessary to screen the samples by conventional atomic absorption until an approximate concentration is determined. After determining the approximate concentration, adjust the volume of the sample accordingly. Pipet 15 ml of concentrated HCl into each tube. Add 1 ml of 30 percent KI solution. Place the reaction tube into a 50 °C (120 °F) water bath for 5 minutes. Cool to room temperature. Connect the reaction tube to the vapor generator assembly. When the instrument response has returned to baseline, inject 5.0 ml of 5 percent NaBH4, and integrate the resulting spectrophotometer signal over a 30-second time period.
11.4.1.1.2 Graphite Furnace Procedure. Dilute the digested sample so that a 5 ml aliquot contains less than 1.5 µg of arsenic. Pipet 5 ml of this digested solution into a 10-ml volumetric flask. Add 1 ml of the 1 percent nickel nitrate solution, 0.5 ml of 50 percent HNO3, and 1 ml of the 3 percent hydrogen peroxide and dilute to 10 ml with water. The sample is now ready for analysis.
11.4.1.2 Run a blank (0.8 N HNO3) and standard at least after every five samples to check the spectrophotometer calibration. The peak height of the blank must pass through a point no further from the origin than ±2 percent of the recorder full scale. The difference between the measured concentration of the standard (the product of the corrected average peak height and the reciprocal of the least squares slope) and the actual concentration of the standard must be less than 7 percent, or recalibration of the analyzer is required.
11.4.1.3 Determine the arsenic concentration in the filter blank (i.e., the average of the two blank values from each lot).
11.4.2 Container Number 3 (Silica Gel). This step may be conducted in the field. Weigh the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g; record this weight.
11.5 Check for matrix effects on the arsenic results. Same as Method 12, Section 11.5.
12.0 Data Analysis and Calculations 12.1 Nomenclature Bws = Water in the gas stream, proportion by volume. Ca = Concentration of arsenic as read from the standard curve, µg/ml. Cs = Arsenic concentration in stack gas, dry basis, converted to standard conditions, g/dsm 3 (gr/dscf). Ea = Arsenic mass emission rate, g/hr (lb/hr). Fd = Dilution factor (equals 1 if the sample has not been diluted). I = Percent of isokinetic sampling. mbi = Total mass of all four impingers and contents before sampling, g. mfi = Total mass of all four impingers and contents after sampling, g. mn = Total mass of arsenic collected in a specific part of the sampling train, µg. mt = Total mass of arsenic collected in the sampling train, µg. Tm = Absolute average dry gas meter temperature (see Figure 108-2), °K (°R). Vm = Volume of gas sample as measured by the dry gas meter, dry basis, m 3 (ft 3). Vm(std) = Volume of gas sample as measured by the dry gas meter, corrected to standard conditions, m 3 (ft 3). Vn = Volume of solution in which the arsenic is contained, ml. Vw(std) = Volume of water vapor collected in the sampling train, corrected to standard conditions, m 3 (ft 3). ΔH = Average pressure differential across the orifice meter (see Figure 108-2), mm H2O (in. H2O).12.2 Average Dry Gas Meter Temperatures (Tm) and Average Orifice Pressure Drop (ΔH). See data sheet (Figure 108-2).
12.3 Dry Gas Volume. Using data from this test, calculate Vm(std) according to the procedures outlined in Method 5, Section 12.3.
12.4 Volume of Water Vapor.
Where: K2 = 0.001334 m 3/g for metric units. = 0.047012 ft 3/g for English units.12.5 Moisture Content.
12.6 Amount of Arsenic Collected.
12.6.1 Calculate the amount of arsenic collected in each part of the sampling train, as follows:
12.6.2 Calculate the total amount of arsenic collected in the sampling train as follows:
12.7 Calculate the arsenic concentration in the stack gas (dry basis, adjusted to standard conditions) as follows:
Where: K3 = 10−6 g/µg for metric units = 1.54 × 10−5 gr/µg for English units12.8 Stack Gas Velocity and Volumetric Flow Rate. Calculate the average stack gas velocity and volumetric flow rate using data obtained in this method and the equations in Sections 12.2 and 12.3 of Method 2.
12.9 Pollutant Mass Rate. Calculate the arsenic mass emission rate as follows:
12.10 Isokinetic Variation. Same as Method 5, Section 12.11.
13.0 Method Performance13.1 Sensitivity. The lower limit of flame AAS 10 µg As/ml. The analytical procedure includes provisions for the use of a graphite furnace or vapor generator for samples with a lower arsenic concentration.
14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 Alternative Procedures16.1 Inductively coupled plasma-atomic emission spectrometry (ICP-AES) Analysis. ICP-AES may be used as an alternative to atomic absorption analysis provided the following conditions are met:
16.1.1 Sample collection, sample preparation, and analytical preparation procedures are as defined in the method except as necessary for the ICP-AES application.
16.1.2 Quality Assurance/Quality Control procedures, including audit material analysis, are conducted as prescribed in the method. The QA acceptance conditions must be met.
16.1.3 The limit of quantitation for the ICP-AES must be demonstrated and the sample concentrations reported should be no less than two times the limit of quantitation. The limit of quantitation is defined as ten times the standard deviation of the blank value. The standard deviation of the blank value is determined from the analysis of seven blanks. It has been reported that for mercury and those elements that form hydrides, a continuous-flow generator coupled to an ICP-AES offers detection limits comparable to cold vapor atomic absorption.
16.2 Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) Analysis. ICP-MS may be used as an alternative to atomic absorption analysis.
16.3 Cold Vapor Atomic Fluorescence Spectrometry (CVAFS) Analysis. CVAFS may be used as an alternative to atomic absorption analysis.
17.0 References.Same as References 1 through 9 of Method 5, Section 17.0, with the addition of the following:
1. Perkin Elmer Corporation. Analytical Methods for Atomic Absorption Spectrophotometry. 303-0152. Norwalk, Connecticut. September 1976. pp. 5-6.
2. Standard Specification for Reagent Water. In: Annual Book of American Society for Testing and Materials Standards. Part 31: Water, Atmospheric Analysis. American Society for Testing and Materials. Philadelphia, PA. 1974. pp. 40-42.
3. Stack Sampling Safety Manual (Draft). U.S. Environmental Protection Agency, Office of Air Quality Planning and Standard, Research Triangle Park, NC. September 1978.
18.0 Tables, Diagrams, Flowcharts, and Validation Data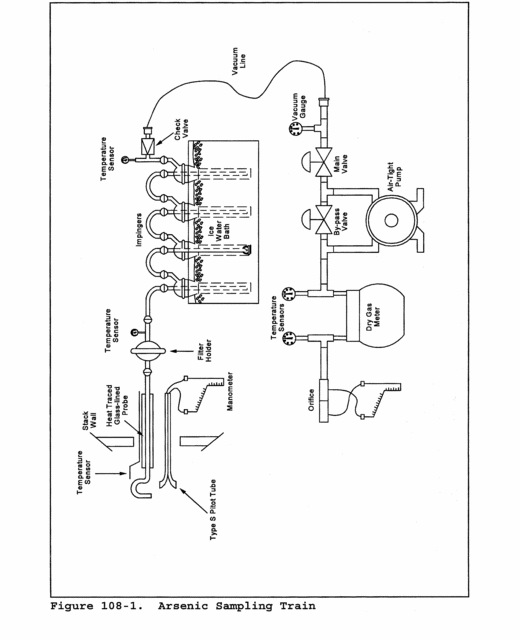
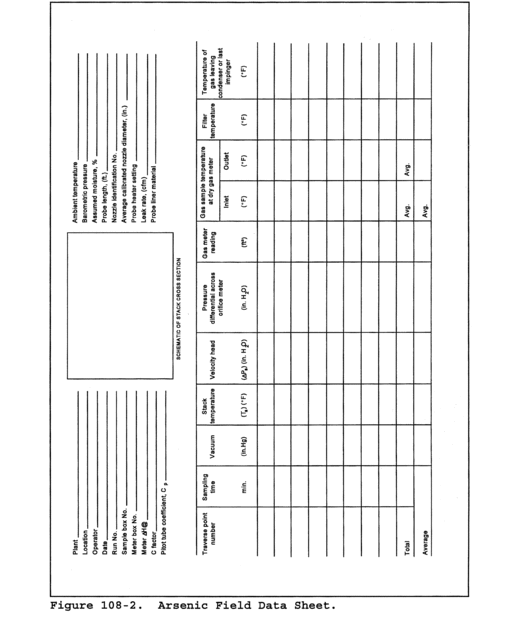 Method 108A -
Determination of Arsenic Content in Ore Samples From Nonferrous
Smelters Note:
Method 108A -
Determination of Arsenic Content in Ore Samples From Nonferrous
Smelters Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in appendix A to 40 CFR part 60. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of Method 12.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Arsenic compounds as arsenic (As) | 7440-38-2 | Lower limit 10 µg/ml or less. |
1.2 Applicability. This method applies to the determination of inorganic As content of process ore and reverberatory matte samples from nonferrous smelters and other sources as specified in an applicable subpart of the regulations.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of MethodArsenic bound in ore samples is liberated by acid digestion and analyzed by flame atomic absorption spectrophotometry (AAS).
3.0 Definitions [Reserved] 4.0 InterferencesAnalysis for As by flame AAS is sensitive to the chemical composition and to the physical properties (e.g., viscosity, pH) of the sample. The analytical procedure includes a check for matrix effects (section 11.5).
5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures that prevent chemical splashes are recommended. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrochloric Acid (HCl). Highly corrosive liquid with toxic vapors. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Hydrofluoric Acid (HF). Highly corrosive to eyes, skin, nose, throat, and lungs. Reaction to exposure may be delayed by 24 hours or more. Provide ventilation to limit exposure.
5.2.3 Hydrogen Peroxide (H2O2). Very harmful to eyes. 30% H2O2 can burn skin, nose, and lungs.
5.2.4 Nitric Acid (HNO3). Highly corrosive to eyes, skin, nose, and lungs. Vapors are highly toxic and can cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.5 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
6.0 Equipment and Supplies6.1 Sample Collection and Preparation. The following items are required for sample collection and preparation:
6.1.1 Parr Acid Digestion Bomb. Stainless steel with vapor-tight Teflon cup and cover.
6.1.2 Volumetric Pipets. 2- and 5-ml sizes.
6.1.3 Volumetric Flask. 50-ml polypropylene with screw caps, (one needed per standard).
6.1.4 Funnel. Polyethylene or polypropylene.
6.1.5 Oven. Capable of maintaining a temperature of approximately 105 °C (221 °F).
6.1.6 Analytical Balance. To measure to within 0.1 mg.
6.2 Analysis. The following items are required for analysis:
6.2.1 Spectrophotometer and Recorder. Equipped with an electrodeless discharge lamp and a background corrector to measure absorbance at 193.7 nm. For measuring samples having less than 10 µg As/ml, use a graphite furnace or vapor generator accessory. The recorder shall match the output of the spectrophotometer.
6.2.2 Volumetric Flasks. Class A, 50-ml (one needed per sample and blank), 500-ml, and 1-liter.
6.2.3 Volumetric Pipets. Class A, 1-, 5-, 10-, and 25-ml sizes.
7.0 Reagents and Standards.Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection and Preparation. The following reagents are required for sample collection and preparation:
7.1.1 Water. Deionized distilled to meet ASTM D 1193-77 or 91 Type 3 (incorporated by reference - See § 61.18). When high concentrations of organic matter are not expected to be present, the KMnO4 test for oxidizable organic matter may be omitted. Use in all dilutions requiring water.
7.1.2 Nitric Acid Concentrated.
7.1.3 Nitric Acid, 0.5 N. In a 1-liter volumetric flask containing water, add 32 ml of concentrated HNO3 and dilute to volume with water.
7.1.4 Hydrofluoric Acid, Concentrated.
7.1.5 Potassium Chloride (KCl) Solution, 10 percent weight by volume (W/V). Dissolve 10 g KCl in water, add 3 ml concentrated HNO3, and dilute to 100 ml.
7.1.6 Filter. Teflon filters, 3-micron porosity, 47-mm size. (Available from Millipore Co., type FS, Catalog Number FSLW04700.)
7.1.7 Sodium Borohydride (NaBH4), 5 Percent (W/V). Dissolve 50.0 g of NaBH4 in about 500 ml of 0.1 N NaOH in a 1-liter volumetric flask. Then, dilute to exactly 1.0 liter with 0.1 N NaOH.
7.1.8 Nickel Nitrate, 5 Percent Ni (W/V). Dissolve 24.780 g of nickel nitrate hexahydrate [Ni(NO3)2 6H2O] in water in a 100-ml volumetric flask, and dilute to 100 ml with water.
7.1.9 Nickel Nitrate, 1 Percent Ni (W/V). Pipet 20 ml of 5 percent nickel nitrate solution into a 100-ml volumetric flask, and dilute to 100 ml with water.
7.2 Analysis. The following reagents and standards are required for analysis:
7.2.2 Sodium Hydroxide, 0.1 N. Dissolve 2.00 g of NaOH in water in a 500-ml volumetric flask. Dilute to volume with water.
7.2.3 Nitric Acid, 0.5 N. Same as in Section 7.1.3.
7.2.4 Potassium Chloride Solution, 10 percent. Same as in Section 7.1.5.
7.2.5 Hydrochloric Acid, Concentrated.
7.2.6 Potassium Iodide (KI), 30 Percent (W/V). Dissolve 300 g of KI in about 500 ml of water in a 1-liter volumetric flask. Then, dilute to exactly 1.0 liter with water.
7.2.7 Hydrogen Peroxide, 3 Percent by Volume. Pipet 50 ml of 30 percent H2O2 into a 500-ml volumetric flask, and dilute to exactly 500 ml with water.
7.2.8 Stock Arsenic Standard, 1 mg As/ml. Dissolve 1.3203 g of primary grade As2O3 in 20 ml of 0.1 N NaOH. Slowly add 30 ml of concentrated HNO3, and heat in an oven at 105 °C (221 °F) for 2 hours. Allow to cool, and dilute to 1 liter with deionized distilled water.
7.2.9 Nitrous Oxide. Suitable quality for AAS analysis.
7.2.10 Acetylene. Suitable quality for AAS analysis.
7.2.11 Quality Assurance Audit Samples. When making compliance determinations, and upon availability, audit samples may be obtained from the appropriate EPA regional Office or from the responsible enforcement authority.
Note:The responsible enforcement authority should be notified at least 30 days prior to the test date to allow sufficient time for sample delivery.
8.0 Sample Collection, Preservation, Transport, and Storage8.1 Sample Collection. A sample that is representative of the ore lot to be tested must be taken prior to analysis. (A portion of the samples routinely collected for metals analysis may be used provided the sample is representative of the ore being tested.)
8.2 Sample Preparation. The sample must be ground into a finely pulverized state.
9.0 Quality Control| Section | Quality control measure | Effect |
|---|---|---|
| 10.2 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
| 11.5 | Check for matrix effects | Eliminate matrix effects. |
Maintain a laboratory log of all calibrations.
10.1 Preparation of Standard Solutions. Pipet 1, 5, 10, and 25 ml of the stock As solution into separate 100-ml volumetric flasks. Add 10 ml KCl solution and dilute to the mark with 0.5 N HNO3. This will give standard concentrations of 10, 50, 100, and 250 µg As/ml. For low-level arsenic samples that require the use of a graphite furnace or vapor generator, follow the procedures in Section 11.3:1. Dilute 10 ml of KCl solution to 100 ml with 0.5 N HNO3 and use as a reagent blank.
10.2 Calibration Curve. Analyze the reagent blank and each standard solution according to the procedures outlined in Section 11.3. Repeat this procedure on each standard solution until two consecutive peaks agree within 3 percent of their average value. Subtract the average peak height (or peak area) of the blank - which must be less than 2 percent of recorder full scale - from the averaged peak heights of each standard solution. If the blank absorbance is greater than 2 percent of full-scale, the probable cause is Hg contamination of a reagent or carry-over of As from a previous sample. Prepare the calibration curve by plotting the corrected peak height of each standard solution versus the corresponding final total As weight in the solution.
10.3 Spectrophotometer Calibration Quality Control. Calculate the least squares slope of the calibration curve. The line must pass through the origin or through a point no further from the origin than ±2 percent of the recorder full scale. Multiply the corrected peak height by the reciprocal of the least squares slope to determine the distance each calibration point lies from the theoretical calibration line. The difference between the calculated concentration values and the actual concentrations must be less than 7 percent for all standards.
Note:For instruments equipped with direct concentration readout devices, preparation of a standard curve will not be necessary. In all cases, follow calibration and operational procedures in the manufacturer's instruction manual.
11.0 Analytical Procedure11.1 Sample Preparation. Weigh 50 to 500 mg of finely pulverized sample to the nearest 0.1 mg. Transfer the sample into the Teflon cup of the digestion bomb, and add 2 ml each of concentrated HNO3 and HF. Seal the bomb immediately to prevent the loss of any volatile arsenic compounds that may form. Heat in an oven at 105 °C (221 °F) for 2 hours. Remove the bomb from the oven and allow to cool. Using a Teflon filter, quantitatively filter the digested sample into a 50-ml polypropylene volumetric flask. Rinse the bomb three times with small portions of 0.5 N HNO3, and filter the rinses into the flask. Add 5 ml of KCl solution to the flask, and dilute to 50 ml with 0.5 N HNO3.
11.2 Spectrophotometer Preparation.
11.2.1 Turn on the power; set the wavelength, slit width, and lamp current. Adjust the background corrector as instructed by the manufacturer's manual for the particular atomic absorption spectrophotometer. Adjust the burner and flame characteristics as necessary.
11.2.2 Develop a spectrophotometer calibration curve as outlined in Sections 10.2 and 10.3.
11.3 Arsenic Determination. Analyze an appropriately sized aliquot of each diluted sample (from Section 11.1) until two consecutive peak heights agree within 3 percent of their average value. If applicable, follow the procedures outlined in Section 11.3.1. If the sample concentration falls outside the range of the calibration curve, make an appropriate dilution with 0.5 N HNO3 so that the final concentration falls within the range of the curve. Using the calibration curve, determine the As concentration in each sample.
Note:Because instruments vary between manufacturers, no detailed operating instructions will be given here. Instead, the instrument manufacturer's detailed operating instructions should be followed.
11.3.1 Arsenic Determination at Low Concentration. The lower limit of flame AAS is 10 µg As/ml. If the arsenic concentration of any sample is at a lower level, use the vapor generator or graphite furnace which is available as an accessory component. Flame, graphite furnace, or vapor generators may be used for samples whose concentrations are between 10 and 30 µg/ml. Follow the manufacturer's instructions in the use of such equipment.
11.3.1.1 Vapor Generator Procedure. Place a sample containing between 0 and 5 µg of arsenic in the reaction tube, and dilute to 15 ml with water. Since there is some trial and error involved in this procedure, it may be necessary to screen the samples by conventional AAS until an approximate concentration is determined. After determining the approximate concentration, adjust the volume of the sample accordingly. Pipet 15 ml of concentrated HCl into each tube. Add 1 ml of 30 percent KI solution. Place the reaction tube into a 50 °C (120 °F) water bath for 5 minutes. Cool to room temperature. Connect the reaction tube to the vapor generator assembly. When the instrument response has returned to baseline, inject 5.0 ml of 5 percent NaBH4 and integrate the resulting spectrophotometer signal over a 30-second time period.
11.3.1.2 Graphite Furnace Procedure. Pipet 5 ml of the digested solution into a 10-ml volumetric flask. Add 1 ml of the 1 percent nickel nitrate solution, 0.5 ml of 50 percent HNO3, and 1 ml of the 3 percent H2O2, and dilute to 10 ml with water. The sample is now ready to inject in the furnace for analysis.
11.4 Run a blank and standard at least after every five samples to check the spectrophotometer calibration. The peak height of the blank must pass through a point no further from the origin than ±2 percent of the recorder full scale. The difference between the measured concentration of the standard (the product of the corrected average peak height and the reciprocal of the least squares slope) and the actual concentration of the standard must be less than 7 percent, or recalibration of the analyzer is required.
11.5 Mandatory Check for Matrix Effects on the Arsenic Results. Same as Method 12, Section 11.5.
12.0 Data Analysis and Calculations12.1 Calculate the percent arsenic in the ore sample as follows:
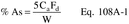 Where: Ca
= Concentration of As as read from the standard curve, µg/ml. Fd =
Dilution factor (equals to 1 if the sample has not been diluted). W
= Weight of ore sample analyzed, mg. 5 = (50 ml sample “ 100)/(10 3
µg/mg). 13.0 Method Performance
Where: Ca
= Concentration of As as read from the standard curve, µg/ml. Fd =
Dilution factor (equals to 1 if the sample has not been diluted). W
= Weight of ore sample analyzed, mg. 5 = (50 ml sample “ 100)/(10 3
µg/mg). 13.0 Method Performance
13.1 Sensitivity. The lower limit of flame AAS is 10 µg As/ml. The analytical procedure includes provisions for the use of a graphite furnace or vapor generator for samples with a lower arsenic concentration.
14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 Alternative Procedures16.1 Alternative Analyzer. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) may be used as an alternative to atomic absorption analysis provided the following conditions are met:
16.1.1 Sample collection, sample preparation, and analytical preparation procedures are as defined in the method except as necessary for the ICP-AES application.
16.1.2 Quality Assurance/Quality Control procedures, including audit material analysis, are conducted as prescribed in the method. The QA acceptance conditions must be met.
16.1.3 The limit of quantitation for the ICP-AES must be demonstrated and the sample concentrations reported should be no less than two times the limit of quantitation. The limit of quantitation is defined as ten times the standard deviation of the blank value. The standard deviation of the blank value is determined from the analysis of seven blanks. It has been reported that for mercury and those elements that form hydrides, a continuous-flow generator coupled to an ICP-AES offers detection limits comparable to cold vapor atomic absorption.
17.0 ReferencesSame as References 1 through 9 of Section 17.0 of Method 5, with the addition of the following:
1. Perkin Elmer Corporation. Analytical Methods of Atomic Absorption Spectrophotometry. 303-0152. Norwalk, Connecticut. September 1976. pp 5-6.
2. Ringwald, D. Arsenic Determination on Process Materials from ASARCO's Copper Smelter in Tacoma, Washington. Unpublished Report. Prepared for Emission Measurement Branch, Emission Standards and Engineering Division, U.S. Environmental Protection Agency, Research Triangle Park, North Carolina. August 1980. 35 pp.
3. Stack Sampling Safety Manual (Draft). U.S. Environmental Protection Agency, Office of Air Quality Planning and Standard, Research Triangle Park, NC. September 1978.
18.0 Tables, Diagrams, Flowcharts, and Validation Data. [Reserved] Method 108B - Determination of Arsenic Content in Ore Samples From Nonferrous Smelters Note:This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this appendix and in appendix A to 40 CFR part 60. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 12 and Method 108A.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Arsenic compounds as arsenic (As) | 7440-38-2 | Lower limit 10 µg/ml. |
1.2 Applicability. This method applies to the determination of inorganic As content of process ore and reverberatory matte samples from nonferrous smelters and other sources as specified in an applicable subpart of the regulations. Samples resulting in an analytical concentration greater than 10 µg As/ml may be analyzed by this method. For lower level arsenic samples, Method 108C should be used.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of MethodArsenic bound in ore samples is liberated by acid digestion and analyzed by flame atomic absorption spectrophotometry (AAS).
3.0 Definitions [Reserved] 4.0 InterferencesAnalysis for As by flame AAS is sensitive to the chemical composition and to the physical properties (e.g., viscosity, pH) of the sample. The analytical procedure includes a check for matrix effects (Section 11.4).
5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures that prevent chemical splashes are recommended. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrochloric acid (HCl). Highly corrosive liquid with toxic vapors. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Hydrofluoric Acid (HF). Highly corrosive to eyes, skin, nose, throat, and lungs. Reaction to exposure may be delayed by 24 hours or more. Provide ventilation to limit exposure.
5.2.3 Nitric Acid (HNO3). Highly corrosive to eyes, skin, nose, and lungs. Vapors are highly toxic and can cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.4 Perchloric Acid (HClO4). Corrosive to eyes, skin, nose, and throat. Provide ventilation to limit exposure. Very strong oxidizer. Keep separate from water and oxidizable materials to prevent vigorous evolution of heat, spontaneous combustion, or explosion. Heat solutions containing HClO4 only in hoods specifically designed for HClO4.
6.0 Equipment and Supplies6.1 Sample Preparation. The following items are required for sample preparation:
6.1.1 Teflon Beakers. 150-ml.
6.1.2 Graduated Pipets. 5-ml disposable.
6.1.3 Graduated Cylinder. 50-ml.
6.1.4 Volumetric Flask. 100-ml.
6.1.5 Analytical Balance. To measure within 0.1 mg.
6.1.6 Hot Plate.
6.1.7 Perchloric Acid Fume Hood.
6.2 Analysis. The following items are required for analysis:
6.2.1 Spectrophotometer. Equipped with an electrodeless discharge lamp and a background corrector to measure absorbance at 193.7 nm.
6.2.2 Beaker and Watch Glass. 400-ml.
6.2.3 Volumetric Flask. 1-liter.
6.2.4 Volumetric Pipets. 1-, 5-, 10-, and 25-ml.
7.0 Reagents and StandardsUnless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Preparation. The following reagents are required for sample preparation:
7.1.1 Water. Deionized distilled to meet ASTM D 1193-77 or 91 Type 3 (incorporated by reference - see § 61.18).
7.1.2 Nitric Acid, Concentrated.
7.1.3 Hydrofluoric Acid, Concentrated.
7.1.4 Perchloric Acid, 70 Percent.
7.1.5 Hydrochloric Acid, Concentrated.
7.2 Analysis. The following reagents and standards are required for analysis:
7.2.1 Water. Same as in Section 7.1.1.
7.2.2 Stock Arsenic Standard, 1.0 mg As/ml. Dissolve 1.3203 g of primary grade As203 [dried at 105 °C (221 °F)] in a 400-ml beaker with 10 ml of HNO3 and 5 ml of HCl. Cover with a watch glass, and heat gently until dissolution is complete. Add 10 ml of HNO3 and 25 ml of HClO4, evaporate to strong fumes of HClO4, and reduce to about 20 ml volume. Cool, add 100 ml of water and 100 ml of HCl, and transfer quantitatively to a 1-liter volumetric flask. Dilute to volume with water and mix.
7.2.3 Acetylene. Suitable quality for AAS analysis.
7.2.4 Air. Suitable quality for AAS analysis.
8.0 Sample Collection, Preservation, Transport, and StorageSame as in Method 108A, Sections 8.1 and 8.2.
9.0 Quality Control| Section | Quality control measure | Effect |
|---|---|---|
| 10.2 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
| 11.4 | Check for matrix effects | Eliminate matrix effects. |
Maintain a laboratory log of all calibrations.
10.1 Preparation of Standard Solutions. Pipet 1, 5, 10, and 25 ml of the stock As solution into separate 100-ml volumetric flasks. Add 2 ml of HClO4, 10 ml of HCl, and dilute to the mark with water. This will provide standard concentrations of 10, 50, 100, and 250 µg As/ml.
10.2 Calibration Curve and Spectrophotometer Calibration Quality Control. Same as Method 108A, Sections 10.2 and 10.3
11.0 Analytical Procedure11.1 Sample Preparation. Weigh 100 to 1000 mg of finely pulverized sample to the nearest 0.1 mg. Transfer the sample to a 150-ml Teflon beaker. Dissolve the sample by adding 15 ml of HNO3, 10 ml of HCl, 10 ml of HF, and 10 ml of HClO4 in the exact order as described, and let stand for 10 minutes. In a HClO4 fume hood, heat on a hot plate until 2-3 ml of HClO4 remain, then cool. Add 20 ml of water and 10 ml of HCl. Cover and warm until the soluble salts are in solution. Cool, and transfer quantitatively to a 100-ml volumetric flask. Dilute to the mark with water.
11.2 Spectrophotometer Preparation. Same as in Method 108A, Section 11.2.
11.3 Arsenic Determination. If the sample concentration falls outside the range of the calibration curve, make an appropriate dilution with 2 percent HClO4/10 percent HCl (prepared by diluting 2 ml concentrated HClO4 and 10 ml concentrated HCl to 100 ml with water) so that the final concentration falls within the range of the curve. Using the calibration curve, determine the As concentration in each sample.
Note:Because instruments vary between manufacturers, no detailed operating instructions will be given here. Instead, the instrument manufacturer's detailed operating instructions should be followed.
Run a blank and standard at least after every five samples to check the spectrophotometer calibration. The peak height of the blank must pass through a point no further from the origin than ±2 percent of the recorder full scale. The difference between the measured concentration of the standard (the product of the corrected average peak height and the reciprocal of the least squares slope) and the actual concentration of the standard must be less than 7 percent, or recalibration of the analyzer is required.
11.4 Mandatory Check for Matrix Effects on the Arsenic Results. Same as Method 12, Section 11.5.
12.0 Data Analysis and CalculationsSame as in Method 108A, Section 12.0.
13.0 Method Performance13.1 Sensitivity. The lower limit of flame AAS is 10 µg As/ml.
14.0 Pollution Prevention [Reserved] 15.0 Waste Management [Reserved] 16.0 ReferencesSame as in Method 108A, Section 16.0.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved] Method 108C - Determination of Arsenic Content in Ore Samples From Nonferrous Smelters (Molybdenum Blue Photometric Procedure) Note:This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least Method 108A.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Arsenic compounds as arsenic (As) | 7440-38-2 | Lower limit 0.0002 percent As by weight. |
1.2 Applicability. This method applies to the determination of inorganic As content of process ore and reverberatory matte samples from nonferrous smelters and other sources as specified in an applicable subpart of the regulations.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of MethodArsenic bound in ore samples is liberated by acid digestion and analyzed by the molybdenum blue photometric procedure.
3.0 Definitions. [Reserved] 4.0 Interferences. [Reserved] 5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures that prevent chemical splashes are recommended. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrochloric Acid (HCl). Highly corrosive liquid with toxic vapors. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Hydrofluoric Acid (HF). Highly corrosive to eyes, skin, nose, throat, and lungs. Reaction to exposure may be delayed by 24 hours or more. Provide ventilation to limit exposure.
5.2.3 Nitric Acid (HNO4). Highly corrosive to eyes, skin, nose, and lungs. Vapors are highly toxic and can cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.4 Perchloric Acid (HClO4). Corrosive to eyes, skin, nose, and throat. Provide ventilation to limit exposure. Very strong oxidizer. Keep separate from water and oxidizable materials to prevent vigorous evolution of heat, spontaneous combustion, or explosion. Heat solutions containing HClO4 only in hoods specifically designed for HClO4.
5.2.5 Sulfuric acid (H2SO4). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 3 mg/m 3 will cause lung damage in uninitiated. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0 Equipment and Supplies6.1 Sample Preparation. The following items are required for sample preparation:
6.1.1 Analytical Balance. To measure to within 0.1 mg.
6.1.2 Erlenmeyer Flask. 300-ml.
6.1.3 Hot Plate.
6.1.4 Distillation Apparatus. No. 6, in ASTM E 50-82, 86, or 90 (Reapproved 1995)(incorporated by reference - see § 61.18); detailed in Figure 108C-1.
6.1.5 Graduated Cylinder. 50-ml.
6.1.6 Perchloric Acid Fume Hood.
6.2 Analysis. The following items are required for analysis:
6.2.1 Spectrophotometer. Capable of measuring at 660 nm.
6.2.2 Volumetric Flasks. 50- and 100-ml.
7.0 Reagents and StandardsUnless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Preparation. The following reagents are required for sample preparation:
7.1.1 Water. Deionized distilled to meet ASTM D 1193-77 or 91 Type 3 (incorporated by reference - see § 61.18). When high concentrations of organic matter are not expected to be present, the KMnO4 test for oxidizable organic matter may be omitted. Use in all dilutions requiring water.
7.1.2 Nitric Acid, Concentrated.
7.1.3 Hydrofluoric Acid, Concentrated.
7.1.4 Sulfuric Acid, Concentrated.
7.1.5 Perchloric Acid, 70 Percent.
7.1.6 Hydrochloric Acid, Concentrated.
7.1.7 Dilute Hydrochloric Acid. Add one part concentrated HCl to nine parts water.
7.1.8 Hydrazine Sulfate ((NH2)2·H2SO4).
7.1.9 Potassium Bromide (KBr).
7.1.10 Bromine Water, Saturated.
7.2 Analysis. The following reagents and standards are required for analysis:
7.2.1 Water. Same as in Section 7.1.1.
7.2.2 Methyl Orange Solution, 1 g/liter.
7.2.3 Ammonium Molybdate Solution, 5 g/liter. Dissolve 0.5 g (NH4)Mo7O24·4H2O in water in a 100-ml volumetric flask, and dilute to the mark. This solution must be freshly prepared.
7.2.4 Standard Arsenic Solution, 10 µg As/ml. Dissolve 0.13203 g of As2O3 in 100 ml HCl in a 1-liter volumetric flask. Add 200 ml of water, cool, dilute to the mark with water, and mix. Transfer 100 ml of this solution to a 1-liter volumetric flask, add 40 ml HCl, cool, dilute to the mark, and mix.
7.2.5 Hydrazine Sulfate Solution, 1 g/liter. Dissolve 0.1 g of [(NH2)2·H2SO4] in water, and dilute to 100 ml in a volumetric flask. This solution must be freshly prepared.
7.2.6 Potassium Bromate (KBrO3) Solution, 0.03 Percent Weight by Volume (W/V). Dissolve 0.3 g KBrO3 in water, and dilute to 1 liter with water.
7.2.7 Ammonium Hydroxide (NH4OH), Concentrated.
7.2.8 Boiling Granules.
7.2.9 Hydrochloric Acid, 50 percent by volume. Dilute equal parts concentrated HCl with water.
8.0 Sample Collection, Preservation, Transport, and StorageSame as in Method 108A, Sections 8.1 and 8.2.
9.0 Quality Control| Section | Quality control measure | Effect |
|---|---|---|
| 10.2 | Calibration curve preparation | Ensure linearity of spectrophotometric response to standards. |
Maintain a laboratory log of all calibrations.
10.1 Preparation of Standard Solutions. Transfer 1.0, 2.0, 4.0, 8.0, 12.0, 16.0, and 20.0 ml of standard arsenic solution (10 µg/ml) to each of seven 50-ml volumetric flasks. Dilute to 20 ml with dilute HCl. Add one drop of methyl orange solution and neutralize to the yellow color with dropwise addition of NH4OH. Just bring back to the red color by dropwise addition of dilute HCl, and add 10 ml in excess. Proceed with the color development as described in Section 11.2.
10.2 Calibration Curve. Plot the spectrophotometric readings of the calibration solutions against µg As per 50 ml of solution. Use this curve to determine the As concentration of each sample.
10.3 Spectrophotometer Calibration Quality Control. Calculate the least squares slope of the calibration curve. The line must pass through the origin or through a point no further from the origin than ±2 percent of the recorder full scale. Multiply the corrected peak height by the reciprocal of the least squares slope to determine the distance each calibration point lies from the theoretical calibration line. The difference between the calculated concentration values and the actual concentrations must be less than 7 percent for all standards.
11.0 Analytical Procedure11.1 Sample Preparation.
11.1.1 Weigh 1.0 g of finely pulverized sample to the nearest 0.1 mg. Transfer the sample to a 300 ml Erlenmeyer flask and add 15 ml of HNO3, 4 ml HCl, 2 ml HF, 3 ml HClO4, and 15 ml H2SO4, in the order listed. In a HClO4 fume hood, heat on a hot plate to decompose the sample. Then heat while swirling over an open flame until dense white fumes evolve. Cool, add 15 ml of water, swirl to hydrate the H2SO4 completely, and add several boiling granules. Cool to room temperature.
11.1.2 Add 1 g of KBr, 1 g hydrazine sulfate, and 50 ml HCl. Immediately attach the distillation head with thermometer and dip the side arm into a 50-ml graduated cylinder containing 25 ml of water and 2 ml of bromine water. Keep the graduated cylinder immersed in a beaker of cold water during distillation. Distill until the temperature of the vapor in the flask reaches 107 °C (225 °F). When distillation is complete, remove the flask from the hot plate, and simultaneously wash down the side arm with water as it is removed from the cylinder.
11.1.3 If the expected arsenic content is in the range of 0.0020 to 0.10 percent, dilute the distillate to the 50-ml mark of the cylinder with water, stopper, and mix. Transfer a 5.0-ml aliquot to a 50-ml volumetric flask. Add 10 ml of water and a boiling granule. Place the flask on a hot plate, and heat gently until the bromine is expelled and the color of methyl orange indicator persists upon the addition of 1 to 2 drops. Cool the flask to room temperature. Neutralize just to the yellow color of the indicator with dropwise additions of NH4OH. Bring back to the red color by dropwise addition of dilute HCl, and add 10 ml excess. Proceed with the molybdenum blue color development as described in Section 11.2.
11.1.4 If the expected arsenic content is in the range of 0.0002 to 0.0010 percent As, transfer either the entire initial distillate or the measured remaining distillate from Section 11.1.2 to a 250-ml beaker. Wash the cylinder with two successive portions of concentrated HNO3, adding each portion to the distillate in the beaker. Add 4 ml of concentrated HClO4, a boiling granule, and cover with a flat watch glass placed slightly to one side. Boil gently on a hot plate until the volume is reduced to approximately 10 ml. Add 3 ml of HNO3, and continue the evaporation until HClO4 is refluxing on the beaker cover. Cool briefly, rinse the underside of the watch glass and the inside of the beaker with about 3-5 ml of water, cover, and continue the evaporation to expel all but 2 ml of the HClO4.
Note:If the solution appears cloudy due to a small amount of antimony distilling over, add 4 ml of 50 percent HCl and 5 ml of water, cover, and warm gently until clear. If cloudiness persists, add 5 ml of HNO3 and 2 ml H2SO4. Continue the evaporation of volatile acids to solubilize the antimony until dense white fumes of H2SO4 appear. Retain at least 1 ml of the H2SO4.
11.1.5 To the 2 ml of HClO4 solution or 1 ml of H2SO4 solution, add 15 ml of water, boil gently for 2 minutes, and then cool. Proceed with the molybdenum blue color development by neutralizing the solution directly in the beaker just to the yellow indicator color by dropwise addition of NH4OH. Obtain the red color by dropwise addition of dilute HCl. Transfer the solution to a 50-ml volumetric flask. Rinse the beaker successively with 10 ml of dilute HCl, followed by several small portions of water. At this point the volume of solution in the flask should be no more than 40 ml. Continue with the color development as described in Section 11.2.
11.2 Analysis.
11.2.1 Add 1 ml of KBrO3 solution to the flask and heat on a low-temperature hot plate to about 50 °C (122 °F) to oxidize the arsenic and methyl orange. Add 5.0 ml of ammonium molybdate solution to the warm solution and mix. Add 2.0 ml of hydrazine sulfate solution, dilute until the solution comes within the neck of the flask, and mix. Place the flask in a 400 ml beaker, 80 percent full of boiling water, for 10 minutes. Enough heat must be supplied to prevent the water bath from cooling much below the boiling point upon inserting the volumetric flask. Remove the flask, cool to room temperature, dilute to the mark, and mix.
11.2.2 Transfer a suitable portion of the reference solution to an absorption cell, and adjust the spectrophotometer to the initial setting using a light band centered at 660 nm. While maintaining this spectrophotometer adjustment, take the readings of the calibration solutions followed by the samples.
12.0 Data Analysis and CalculationsSame as in Method 108A, Section 12.0.
13.0 Method Performance. [Reserved] 14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 References1. Ringwald, D. Arsenic Determination on Process Materials from ASARCO's Copper Smelter in Tacoma, Washington. Unpublished Report. Prepared for the Emission Measurement Branch, Technical Support Division, U.S. Environmental Protection Agency, Research Triangle Park, North Carolina. August 1980. 35 pp.
17.0 Tables, Diagrams, Flowcharts, and Validation Data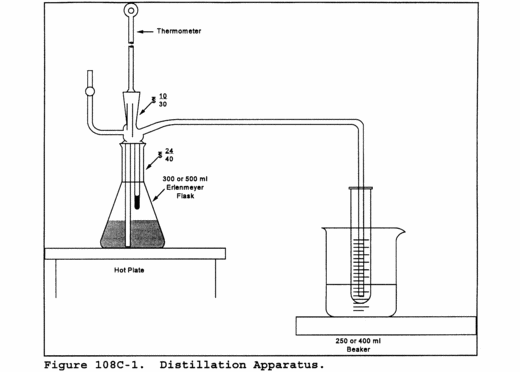 Method 111 -
Determination of Polonium-210 Emissions From Stationary Sources
Note:
Method 111 -
Determination of Polonium-210 Emissions From Stationary Sources
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from methods in appendix A to 40 CFR part 60. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, and Method 5.
1.0 Scope and Application1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Polonium | 7440-08-6 | Not specified. |
1.2 Applicability. This method is applicable for the determination of the polonium-210 content of particulate matter samples collected from stationary source exhaust stacks, and for the use of these data to calculate polonium-210 emissions from individual sources and from all affected sources at a facility.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of MethodA particulate matter sample, collected according to Method 5, is analyzed for polonium-210 content: the polonium-210 in the sample is put in solution, deposited on a metal disc, and the radioactive disintegration rate measured. Polonium in acid solution spontaneously deposits on surfaces of metals that are more electropositive than polonium. This principle is routinely used in the radiochemical analysis of polonium-210. Data reduction procedures are provided, allowing the calculation of polonium-210 emissions from individual sources and from all affected sources at a facility, using data obtained from Methods 2 and 5 and from the analytical procedures herein.
3.0 Definitions [Reserved] 4.0 Interferences [Reserved] 5.0 Safety5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrochloric Acid (HCl). Highly corrosive liquid with toxic vapors. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Hydrofluoric Acid (HF). Highly corrosive to eyes, skin, nose, throat, and lungs. Reaction to exposure may be delayed by 24 hours or more. Provide ventilation to limit exposure.
5.2.3 Nitric Acid (HNO3). Highly corrosive to eyes, skin, nose, and lungs. Vapors cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.4 Perchloric Acid (HClO4). Corrosive to eyes, skin, nose, and throat. Provide ventilation to limit exposure. Keep separate from water and oxidizable materials to prevent vigorous evolution of heat, spontaneous combustion, or explosion. Heat solutions containing HClO4 only in hoods specifically designed for HClO4.
6.0 Equipment and Supplies6.1 Alpha Spectrometry System. Consisting of a multichannel analyzer, biasing electronics, silicon surface barrier detector, vacuum pump and chamber.
6.2 Constant Temperature Bath at 85 °C (185 °F).
6.3 Polished Silver Discs. 3.8 cm diameter, 0.4 mm thick with a small hole near the edge.
6.4 Glass Beakers. 400 ml, 150 ml.
6.5 Hot Plate, Electric.
6.6 Fume Hood.
6.7 Teflon Beakers, 150 ml.
6.8 Magnetic Stirrer.
6.9 Stirring Bar.
6.10 Hooks. Plastic or glass, to suspend plating discs.
6.11 Internal Proportional Counter. For measuring alpha particles.
6.12 Nucleopore Filter Membranes. 25 mm diameter, 0.2 micrometer pore size or equivalent.
6.13 Planchets. Stainless steel, 32 mm diameter with 1.5 mm lip.
6.14 Transparent Plastic Tape. 2.5 cm wide with adhesive on both sides.
6.15 Epoxy Spray Enamel.
6.16 Suction Filter Apparatus. For 25 mm diameter filter.
6.17 Wash Bottles, 250 ml capacity.
6.18 Graduated Cylinder, plastic, 25 ml capacity.
6.19 Volumetric Flasks, 100 ml, 250 ml.
7.0 Reagents and StandardsUnless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Ascorbic Acid.
7.2 Ammonium Hydroxide (NH4OH), 15 M.
7.3 Water. Deionized distilled, to conform to ASTM D 1193-77 or 91 (incorporated by reference - see § 61.18), Type 3. Use in all dilutions requiring water.
7.4 Ethanol (C2H5OH), 95 percent.
7.5 Hydrochloric Acid, 12 M.
7.6 Hydrochloric Acid, 1 M. Dilute 83 ml of the 12 M HCl to 1 liter with distilled water.
7.7 Hydrofluoric Acid, 29 M.
7.8 Hydrofluoric Acid, 3 M. Dilute 52 ml of the 29 M HF to 500 ml with distilled water. Use a plastic graduated cylinder and storage bottle.
7.9 Lanthanum Carrier, 0.1 mg La+3/ml. Dissolve 0.078 gram lanthanum nitrate, La(NO3)3·6H2O in 250 ml of 1 M HCl.
7.10 Nitric Acid, 16 M.
7.11 Perchloric Acid, 12 M.
7.12 Polonium-209 Solution.
7.13 Silver Cleaner. Any mild abrasive commercial silver cleaner.
7.14 Degreaser.
7.15 Standard Solution. Standardized solution of an alpha-emitting actinide element, such as plutonium-239 or americium-241.
8.0 Sample Collection, Preservation, Transport, and Storage. [Reserved] 9.0 Quality Control9.1 General Requirement.
9.1.1 All analysts using this method are required to demonstrate their ability to use the method and to define their respective accuracy and precision criteria.
9.2 Miscellaneous Quality Control Measures| Section | Quality control measure | Effect |
|---|---|---|
| 10.1 | Standardization of alpha spectrometry system | Ensure precision of sample analyses. |
| 10.3 | Standardization of internal proportional counter | Ensure precise sizing of sample aliquot. |
| 11.1, 11.2 | Determination of procedure background and instrument background | Minimize background effects. |
10.1 Standardization of Alpha Spectrometry System.
10.1.1 Add a quantity of the actinide standard solution to a 100 ml volumetric flask so that the final concentration when diluted to a volume of 100 ml will be approximately 1 pCi/ml.
10.1.2 Add 10 ml of 16 M HNO3 and dilute to 100 ml with water.
10.1.3 Add 20 ml of 1 M HCl to each of six 150 ml beakers. Add 1.0 ml of lanthanum carrier, 0.1 mg lanthanum per ml, to the acid solution in each beaker.
10.1.4 Add 1.0 ml of the 1 pCi/ml working solution (from Section 10.1.1) to each beaker. Add 5.0 ml of 3 M HF to each beaker.
10.1.5 Cover beakers and allow solutions to stand for a minimum of 30 minutes. Filter the contents of each beaker through a separate filter membrane using the suction filter apparatus. After each filtration, wash the filter membrane with 10 ml of distilled water and 5 ml of ethanol, and allow the filter membrane to air dry on the filter apparatus.
10.1.6 Carefully remove the filter membrane and mount it, filtration side up, with double-side tape on the inner surface of a planchet. Place planchet in an alpha spectrometry system and count each planchet for 1000 minutes.
10.1.7 Calculate the counting efficiency of the detector for each aliquot of the 1 pCi/ml actinide working solution using Eq. 111-1 in Section 12.2.
10.1.8 Determine the average counting efficiency of the detector, Ec, by calculating the average of the six determinations.
10.2 Preparation of Standardized Solution of Polonium-209.
10.2.1 Add a quantity of the Po-209 solution to a 100 ml volumetric flask so that the final concentration when diluted to a 100 ml volume will be approximately 1 pCi/ml.
10.2.2 Follow the procedures outlined in Sections 10.1.2 through 10.1.6, except substitute 1.0 ml of polonium-209 tracer solution (Section 10.2.1) and 3.0 ml of 15 M ammonium hydroxide for the 1 pCi/ml actinide working solution and the 3 M HF, respectively.
10.2.3 Calculate the activity of each aliquot of the polonium-209 tracer solution using Eq. 111-2 in Section 12.3.
10.2.4 Determine the average activity of the polonium-209 tracer solution, F, by averaging the results of the six determinations.
10.3 Standardization of Internal Proportional Counter
10.3.1 Add a quantity of the actinide standard solution to a 100 ml volumetric flask so that the final concentration when diluted to a 100 ml volume will be approximately 100 pCi/ml.
10.3.2 Follow the procedures outlined in Sections 10.1.2 through 10.1.6, except substitute the 100 pCi/ml actinide working solution for the 1 pCi/ml solution, place the planchet in an internal proportional counter (instead of an alpha spectrometry system), and count for 100 minutes (instead of 1000 minutes).
10.3.3 Calculate the counting efficiency of the internal proportional counter for each aliquot of the 100 pCi/ml actinide working solution using Eq. 111-3 in 12.4.
10.3.4 Determine the average counting efficiency of the internal proportional counter, EI, by averaging the results of the six determinations.
11.0 Analytical Procedure Note:Perform duplicate analyses of all samples, including background counts and Method 5 samples. Duplicate measurements are considered acceptable when the difference between them is less than two standard deviations as described in EPA 600/4-77-001 or subsequent revisions.
11.1 Determination of Procedure Background. Background counts used in all equations are determined by performing the specific analysis required using the analytical reagents only. All procedure background counts and sample counts for the internal proportional counter should utilize a counting time of 100 minutes; for the alpha spectrometry system, 1000 minutes. These background counts should be performed no less frequently than once per 10 sample analyses.
11.2 Determination of Instrument Background. Instrument backgrounds of the internal proportional counter and the alpha spectrometry system should be determined on a weekly basis. Instrument background should not exceed procedure background. If this occurs, it may be due to a malfunction or contamination, and should be corrected before use.
11.4 Sample Preparation. Treat the Method 5 samples [i.e., the glass fiber filter (Container No. 1) and the acetone rinse (Container No. 2)] as follows:
11.4.1 Container No. 1. Transfer the filter and any loose particulate matter from the sample container to a 150-ml Teflon beaker.
11.4.2 Container No. 2. Note the level of liquid in the container, and confirm on the analysis sheet whether leakage occurred during transport. If a noticeable amount of leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results. Transfer the contents to a 400-ml glass beaker. Add polonium-209 tracer solution to the glass beaker in an amount approximately equal to the amount of polonium-210 expected in the total particulate sample. Record the activity of the tracer solution added. Add 16 M nitric acid to the beaker to digest and loosen the residue.
11.4.3 Transfer the contents of the glass beaker to the Teflon beaker containing the glass fiber filter. Rinse the glass beaker with 16 M HNO3. If necessary, reduce the volume in the beaker by evaporation until all of the nitric acid HNO3 from the glass beaker has been transferred to the Teflon beaker.
11.4.4 Add 30 ml of 29 M HF to the Teflon beaker and evaporate to near dryness on a hot plate in a properly operating hood.
Note:Do not allow the residue to go to dryness and overheat; this will result in loss of polonium.
11.4.5 Repeat step 11.4.4 until the filter is dissolved.
11.4.6 Add 100 ml of 16 M HNO3 to the residue in the Teflon beaker and evaporate to near dryness.
Note:Do not allow the residue to go to dryness.
11.4.7 Add 50 ml of 16 M HNO3 and 10 ml of 12 M perchloric acid to the Teflon beaker and heat until dense fumes of perchloric acid are evolved.
11.4.8 Repeat steps 11.4.4 to 11.4.7 as necessary until sample is completely dissolved.
11.4.9 Add 10 ml of 12 M HCl to the Teflon beaker and evaporate to dryness. Repeat additions and evaporations several times.
11.4.10 Transfer the sample to a 250-ml volumetric flask and dilute to volume with 3 M HCl.
11.5 Sample Screening. To avoid contamination of the alpha spectrometry system, check each sample as follows:
11.5.1 Add 20 ml of 1 M HCl, 1 ml of the lanthanum carrier solution (0.1 mg La/ml), a 1 ml aliquot of the sample solution from Section 11.4.10, and 3 ml of 15 M ammonium hydroxide to a 250-ml beaker in the order listed. Allow this solution to stand for a minimum of 30 minutes.
11.5.2 Filter the solution through a filter membrane using the suction filter apparatus. Wash the filter membrane with 10 ml of water and 5 ml of ethanol, and allow the filter membrane to air dry on the filter apparatus.
11.5.3 Carefully remove the filter membrane and mount it, filtration side up, with double-side tape on the inner surface of a planchet. Place the planchet in an internal proportional counter, and count for 100 minutes.
11.5.4 Calculate the activity of the sample using Eq. 111-4 in Section 12.5.
11.5.5 Determine the aliquot volume of the sample solution from Section 11.4.10 to be analyzed for polonium-210, such that the aliquot contains an activity between 1 and 4 picocuries. Use Eq. 111-5 in Section 12.6.
11.6 Preparation of Silver Disc for Spontaneous Electrodeposition.
11.6.1 Clean both sides of the polished silver disc with silver cleaner and with degreaser.
11.6.2 Place disc on absorbent paper and spray one side with epoxy spray enamel. This should be carried out in a well-ventilated area, with the disc lying flat to keep paint on one side only. Allow paint to dry for 24 hours before using disc for deposition.
11.7 Sample Analysis.
11.7.1 Add the aliquot of sample solution from Section 11.4.10 to be analyzed for polonium-210, the volume of which was determined in Section 11.5.5, to a suitable 200-ml container to be placed in a constant temperature bath.
Note:Aliquot volume may require a larger container.
11.7.2 If necessary, bring the volume to 100 ml with 1 M HCl. If the aliquot volume exceeds 100 ml, use total aliquot.
11.7.3 Add 200 mg of ascorbic acid and heat solution to 85 °C (185 °F) in a constant temperature bath.
11.7.4 Suspend a silver disc in the heated solution using a glass or plastic rod with a hook inserted through the hole in the disc. The disc should be totally immersed in the solution, and the solution must be stirred constantly, at all times during the plating operation. Maintain the disc in solution for 3 hours.
11.7.5 Remove the silver disc, rinse with deionized distilled water, and allow to air dry at room temperature.
11.7.6 Place the disc, with deposition side (unpainted side) up, on a planchet and secure with double-side plastic tape. Place the planchet with disc in alpha spectrometry system and count for 1000 minutes.
12.0 Data Analysis and Calculations.12.1 Nomenclature.
A = Picocuries of polonium-210 in the Method 5 sample (from Section 12.8). AA = Picocuries of actinide added. AL = Volume of sample aliquot used, in ml (specified in Section 11.5.1 as 1 ml). AS = Aliquot to be analyzed, in ml. BB = Procedure background counts measured in polonium-209 spectral region. BT = Polonium-209 tracer counts in sample. CT = Total counts in polonium-210 spectral region. D = Decay correction for time “t” (in days) from sample collection to sample counting, given by: D = e−0.005t EC = Average counting efficiency of detector (from Section 10.1.8), as counts per disintegration. ECi = Counting efficiency of the detector for aliquot i of the actinide working solution, counts per disintegration. EI = Average counting efficiency of the internal proportional counter, as determined in Section 10.3.4, counts per disintegration. EIi = Counting efficiency of the internal proportional counter for aliquot i of the 100 pCi/ml actinide working solution, counts per disintegration. EY = The fraction of polonium-209 recovered on the planchet (from Section 12.7). F= Average activity of polonium-209 in sample (from Section 10.2.4), in pCi. Fi = activity of aliquot i of the polonium-209 tracer solution, in pCi. L = Dilution factor (unitless). This is the volume of sample solution prepared (specified as 250 ml in Section 11.1.10) divided by the volume of the aliquot of sample solution analyzed for polonium-210 (from Section 11.7.1). Mi = Phosphorous rock processing rate of the source being tested, during run i, Mg/hr. Mk = Phosphate rock processed annually by source k, in Mg/yr. n = Number of calciners at the elemental phosphorus plant. P = Total activity of sample solution from Section 11.4.10, in pCi (see Eq. 111-4). Qsd = Volumetric flow rate of effluent stream, as determined by Method 2, in dscm/hr. S = Annual polonium-210 emissions from the entire facility, in curies/yr. Vm(std) = Volume of air sample, as determined by Method 5, in dscm. Xk = Emission rate from source k, from Section 12.10, in curies/Mg. 10−12 = Curies per picocurie. 2.22 = Disintegrations per minute per picocurie. 250 = Volume of solution from Section 11.4.10, in ml.12.2 Counting Efficiency. Calculate the counting efficiency of the detector for each aliquot of the 1 pCi/ml actinide working solution using Eq. 111-1.
Where: CB = Background counts in same peak area as CS. CS = Gross counts in actinide peak. T = Counting time in minutes, specified in Section 10.1.6 as 1000 minutes.12.3 Polonium-209 Tracer Solution Activity. Calculate the activity of each aliquot of the polonium-209 tracer solution using Eq. 111-2.
Where: CB = Background counts in the 4.88 MeV region of spectrum the in the counting time T. CS = Gross counts of polonium-209 in the 4.88 MeV region of the spectrum in the counting time T. T = Counting time, specified in Section 10.1.6 as 1000 minutes.12.4 Control Efficiency of Internal Proportional Counter. Calculate the counting efficiency of the internal proportional counter for each aliquot of the 100 pCi/ml actinide working solution using Eq. 111-3.
Where: CB = Gross counts of procedure background. CS = Gross counts of standard. T = Counting time in minutes, specified in Section 10.3.2 as 100 minutes.12.5 Calculate the activity of the sample using Eq. 111-4.
Where: CB = Total counts of procedure background. (See Section 11.1). CS = Total counts of screening sample. T = Counting time for sample and background (which must be equal), in minutes (specified in Section 11.5.3 as 100 minutes).12.6 Aliquot Volume. Determine the aliquot volume of the sample solution from Section 11.4.10 to be analyzed for polonium-210 , such that the aliquot contains an activity between 1 and 4 picocuries using Eq. 111-5.
12.7 Polonium-209 Recovery. Calculate the fraction of polonium-209 recovered on the planchet, EY, using Eq. 111-6.
Where: T = Counting time, specified in Section 11.1 as 1000 minutes.12.8 Polonium-210 Activity. Calculate the activity of polonium-210 in the Method 5 sample (including glass fiber filter and acetone rinse) using Eq. 111-7.
Where: CB = Procedure background counts in polonium-210 spectral region. T = Counting time, specified in Section 11.1 as 1000 minutes for all alpha spectrometry sample and background counts.12.9 Emission Rate from Each Stack.
12.9.1 For each test run, i, on a stack, calculate the measured polonium-210 emission rate, RSi, using Eq. 111-8.
12.9.2 Determine the average polonium-210 emission rate from the stack, RS, by taking the sum of the measured emission rates for all runs, and dividing by the number of runs performed.
12.9.3 Repeat steps 12.9.1 and 12.9.2 for each stack of each calciner.
12.10 Emission Rate from Each Source. Determine the total polonium-210 emission rate, Xk, from each source, k, by taking the sum of the average emission rates from all stacks to which the source exhausts.
12.11 Annual Polonium-210 Emission Rate from Entire Facility. Determine the annual elemental phosphorus plant emissions of polonium-210, S, using Eq. 111-9.
13.0 Method Performance. [Reserved] 14.0 Pollution Prevention. [Reserved] 15.0 Waste Management. [Reserved] 16.0 References1. Blanchard, R.L. “Rapid Determination of Lead-210 and Polonium-210 in Environmental Samples by Deposition on Nickel.” Anal. Chem., 38:189, pp. 189-192. February 1966.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved] Method 114 - Test Methods for Measuring Radionuclide Emissions from Stationary Sources 1. Purpose and BackgroundThis method provides the requirements for: (1) Stack monitoring and sample collection methods appropriate for radionuclides; (2) radiochemical methods which are used in determining the amounts of radionuclides collected by the stack sampling and; (3) quality assurance methods which are conducted in conjunction with these measurements. These methods are appropriate for emissions for stationary sources. A list of references is provided.
Many different types of facilities release radionuclides into air. These radionuclides differ in the chemical and physical forms, half-lives and type of radiation emitted. The appropriate combination of sample extraction, collection and analysis for an individual radionuclide is dependent upon many interrelated factors including the mixture of other radionuclides present. Because of this wide range of conditions, no single method for monitoring or sample collection and analysis of a radionuclide is applicable to all types of facilities. Therefore, a series of methods based on “principles of measurement” are described for monitoring and sample collection and analysis which are applicable to the measurement of radionuclides found in effluent streams at stationary sources. This approach provides the user with the flexibility to choose the most appropriate combination of monitoring and sample collection and analysis methods which are applicable to the effluent stream to be measured.
2. Stack Monitoring and Sample Collection MethodsMonitoring and sample collection methods are described based on “principles of monitoring and sample collection” which are applicable to the measurement of radionuclides from effluent streams at stationary sources. Radionuclides of most elements will be in the particulate form in these effluent streams and can be readily collected using a suitable filter media. Radionuclides of hydrogen, oxygen, carbon, nitrogen, the noble gases and in some circumstances iodine will be in the gaseous form. Radionuclides of these elements will require either the use of an in-line or off-line monitor to directly measure the radionuclides, or suitable sorbers, condensers or bubblers to collect the radionuclides.
2.1 Radionuclides as Particulates. The extracted effluent stream is passed through a filter media to remove the particulates. The filter must have a high efficiency for removal of sub-micron particles. The guidance in ANSI/HPS N13.1-1999 (section 6.6.2 Filter media) shall be followed in using filter media to collect particulates (incorporated by reference - see § 61.18 of this part).
2.2 Radionuclides as Gases.
2.2.1 The Radionuclide Tritium (H-3). Tritium in the form of water vapor is collected from the extracted effluent sample by sorption, condensation or dissolution techniques. Appropriate collectors may include silica gel, molecular sieves, and ethylene glycol or water bubblers.
Tritium in the gaseous form may be measured directly in the sample stream using Method B-1, collected as a gas sample or may be oxidized using a metal catalyst to tritiated water and collected as described above.
2.2.2 Radionuclides of Iodine. Iodine is collected from an extracted sample by sorption or dissolution techniques. Appropriate collectors may include charcoal, impregnated charcoal, metal zeolite and caustic solutions.
2.2.3 Radionuclides of Argon, Krypton and Xenon. Radionuclides of these elements are either measured directly by an in-line or off-line monitor, or are collected from the extracted sample by low temperature sorption techniques, Appropriate sorbers may include charcoal or metal zeolite.
2.2.4 Radionuclides of Oxygen, Carbon, Nitrogen and Radon. Radionuclides of these elements are measured directly using an in-line or off-line monitor. Radionuclides of carbon in the form of carbon dioxide may be collected by dissolution in caustic solutions.
2.3 Definition of Terms
In-line monitor means a continuous measurement system in which the detector is placed directly in or adjacent to the effluent stream. This may involve either gross radioactivity measurements or specific radionuclide measurements. Gross measurements shall be made in conformance with the conditions specified in Methods A-4, B-2 and G-4.
Off-line monitor means a measurement system in which the detector is used to continuously measure an extracted sample of the effluent stream. This may involve either gross radioactivity measurements or specific radionuclide measurements. Gross measurements shall be made in conformance with the conditions specified in Methods A-4, B-2 and G-4.
Sample collection means a procedure in which the radionuclides are removed from an extracted sample of the effluent using a collection media. These collection media include filters, absorbers, bubblers and condensers. The collected sample is analyzed using the methods described in Section 3.
3. Radionuclide Analysis MethodsA series of methods based on “principles of measurement” are described which are applicable to the analysis of radionuclides collected from airborne effluent streams at stationary sources. These methods are applicable only under the conditions stated and within the limitations described. Some methods specify that only a single radionuclide be present in the sample or the chemically separated sample. This condition should be interpreted to mean that no other radionuclides are present in quantities which would interfere with the measurement.
Also identified (Table 1) are methods for a selected list of radionuclides. The listed radionuclides are those which are most commonly used and which have the greatest potential for causing dose to members of the public. Use of methods based on principles of measurement other than those described in this section must be approved in advance of use by the Administrator. For radionuclides not listed in Table 1, any of the described methods may be used provided the user can demonstrate that the applicability conditions of the method have been met.
The type of method applicable to the analysis of a radionuclide is dependent upon the type of radiation emitted, i.e., alpha, beta or gamma. Therefore, the methods described below are grouped according to principles of measurements for the analysis of alpha, beta and gamma emitting radionuclides.
3.1 Methods for Alpha Emitting Radionuclides
3.1.1 Method A-1, Radiochemistry-Alpha Spectrometry.
Principle: The element of interest is separated from other elements, and from the sample matrix using radiochemical techniques. The procedure may involve precipitation, ion exchange, or solvent extraction. Carriers (elements chemically similar to the element of interest) may be used. The element is deposited on a planchet in a very thin film by electrodeposition or by coprecipitation on a very small amount of carrier, such as lanthanum fluoride. The deposited element is then counted with an alpha spectrometer. The activity of the nuclide of interest is measured by the number of alpha counts in the appropriate energy region. A correction for chemical yield and counting efficiency is made using a standardized radioactive nuclide (tracer) of the same element. If a radioactive tracer is not available for the element of interest, a predetermined chemical yield factor may be used.
Applicability: This method is applicable for determining the activity of any alpha-emitting radionuclide, regardless of what other radionuclides are present in the sample provided the chemical separation step produces a very thin sample and removes all other radionuclides which could interfere in the spectral region of interest. APHA-605(2), ASTM-D-3972(13).
3.1.2 Method A-2, Radiochemistry-Alpha Counting.
Principle: The element of interest is separated from other elements, and from the sample matrix using radiochemistry. The procedure may involve precipitation, ion exchange, or solvent extraction. Carriers (elements chemically similar to the element of interest) may be used. The element is deposited on a planchet in a thin film and counted with an alpha counter. A correction for chemical yield (if necessary) is made. The alpha count rate measures the total activity of all emitting radionuclides of the separated element.
Applicability: This method is applicable for the measurement of any alpha-emitting radionuclide, provided no other alpha emitting radionuclide is present in the separated sample. It may also be applicable for determining compliance, when other radionuclides of the separated element are present, provided that the calculated emission rate is assigned to the radionuclide which could be present in the sample that has the highest dose conversion factor. IDO-12096(18).
3.1.3 Method A-3, Direct Alpha Spectrometry.
Principle: The sample, collected on a suitable filter, is counted directly on an alpha spectrometer. The sample must be thin enough and collected on the surface of the filter so that any absorption of alpha particle energy in the sample or the filter, which would degrade the spectrum, is minimal.
Applicability: This method is applicable to simple mixtures of alpha emitting radionuclides and only when the amount of particulates collected on the filter paper are relatively small and the alpha spectra is adequately resolved. Resolutions should be 50 keV (FWHM) or better, ASTM-D-3084(16).
3.1.4 Method A-4, Direct Alpha Counting (Gross alpha determination).
Principle: The sample, collected on a suitable filter, is counted with an alpha counter. The sample must be thin enough so that self-absorption is not significant and the filter must be of such a nature that the particles are retained on the surface.
Applicability: Gross alpha determinations may be used to measure emissions of specific radionuclides only (1) when it is known that the sample contains only a single radionuclide, or the identity and isotopic ratio of the radionuclides in the sample are well-known, and (2) measurements using either Method A-1, A-2 or A-5 have shown that this method provides a reasonably accurate measurement of the emission rate. Gross alpha measurements are applicable to unidentified mixtures of radionuclides only for the purposes and under the conditions described in section 3.7. APHA-601(3), ASTM-D-1943(10).
3.1.5 Method A-5, Chemical Determination of Uranium.
Principle: Uranium may be measured chemically by either colorimetry or fluorometry. In both procedures, the sample is dissolved, the uranium is oxidized to the hexavalent form and extracted into a suitable solvent. Impurities are removed from the solvent layer. For colorimetry, dibenzoylmethane is added, and the uranium is measured by the absorbance in a colorimeter. For fluorometry, a portion of the solution is fused with a sodium fluoride-lithium fluoride flux and the uranium is determined by the ultraviolet activated fluorescence of the fused disk in a fluorometer.
Applicability: This method is applicable to the measurements of emission rates of uranium when the isotopic ratio of the uranium radionuclides is well known. ASTM-E-318(15), ASTM-D-2907(14).
3.1.6 Method A-6, Radon-222 - Continuous Gas Monitor.
Principle: Radon-222 is measured directly in a continuously extracted sample stream by passing the air stream through a calibrated scintillation cell. Prior to the scintillation cell, the air stream is treated to remove particulates and excess moisture. The alpha particles from radon-222 and its decay products strike a zinc sulfide coating on the inside of the scintillation cell producing light pulses. The light pulses are detected by a photomultiplier tube which generates electrical pulses. These pulses are processed by the system electronics and the read out is in pCi/l of radon-222.
Applicability: This method is applicable to the measurement of radon-222 in effluent streams which do not contain significant quantities of radon-220. Users of this method should calibrate the monitor in a radon calibration chamber at least twice per year. The background of the monitor should also be checked periodically by operating the instrument in a low radon environment. EPA 520/1-89-009(24).
3.1.7 Method A-7, Radon-222-Alpha Track Detectors
Principle: Radon-222 is measured directly in the effluent stream using alpha track detectors (ATD). The alpha particles emitted by radon-222 and its decay products strike a small plastic strip and produce submicron damage tracks. The plastic strip is placed in a caustic solution that accentuates the damage tracks which are counted using a microscope or automatic counting system. The number of tracks per unit area is correlated to the radon concentration in air using a conversion factor derived from data generated in a radon calibration facility.
Applicability: Prior approval from EPA is required for use of this method. This method is only applicable to effluent streams which do not contain significant quantities of radon-220, unless special detectors are used to discriminate against radon-220. This method may be used only when ATDs have been demonstrated to produce data comparable to data obtained with Method A-6. Such data should be submitted to EPA when requesting approval for the use of this method. EPA 520/1-89-009(24).
3.2 Methods for Gaseous Beta Emitting Radionuclides.
3.2.1 Method B-1, Direct Counting in Flow-Through Ionization Chambers.
Principle: An ionization chamber containing a specific volume of gas which flows at a given flow rate through the chamber is used. The sample (effluent stream sample) acts as the counting gas for the chamber. The activity of the radionuclide is determined from the current measured in the ionization chamber.
Applicability: This method is applicable for measuring the activity of a gaseous beta-emitting radionuclide in an effluent stream that is suitable as a counting gas, when no other beta-emitting nuclides are present. DOE/EP-0096(17), NCRP-58(23).
3.2.2 Method B-2, Direct Counting With In-line or Off-line Beta Detectors.
Principle: The beta detector is placed directly in the effluent stream (in-line) or an extracted sample of the effluent stream is passed through a chamber containing a beta detector (off-line). The activities of the radionuclides present in the effluent stream are determined from the beta count rate, and a knowledge of the radionuclides present and the relationship of the gross beta count rate and the specific radionuclide concentration.
Applicability: This method is applicable only to radionuclides with maximum beta particle energies greater then 0.2 MeV. This method may be used to measure emissions of specific radionuclides only when it is known that the sample contains only a single radionuclide or the identity and isotopic ratio of the radionuclides in the effluent stream are well known. Specific radionuclide analysis of periodic grab samples may be used to identify the types and quantities of radionuclides present and to establish the relationship between specific radionuclide analyses and gross beta count rates.
This method is applicable to unidentified mixtures of gaseous radionuclides only for the purposes and under the conditions described in section 3.7.
3.3 Methods for Non-Gaseous Beta Emitting Radionuclides.
3.3.1 Method B-3, Radiochemistry-Beta Counting.
Principle: The element of interest is separated from other elements, and from the sample matrix by radiochemistry. This may involve precipitation, distillation, ion exchange, or solvent extraction. Carriers (elements chemically similar to the element of interest) may be used. The element is deposited on a planchet, and counted with a beta counter. Corrections for chemical yield, and decay (if necessary) are made. The beta count rate determines the total activity of all radionuclides of the separated element. This method may also involve the radiochemical separation and counting of a daughter element, after a suitable period of ingrowth, in which case it is specific for the parent nuclide.
Applicability: This method is applicable for measuring the activity of any beta-emitting radionuclide, with a maximum energy greater than 0.2 MeV, provided no other radionuclide is present in the separated sample. APHA-608(5).
3.3.2 Method B-4, Direct Beta Counting (Gross beta determination).
Principle: The sample, collected on a suitable filter, is counted with a beta counter. The sample must be thin enough so that self-absorption corrections can be made.
Applicability: Gross beta measurements are applicable only to radionuclides with maximum beta particle energies greater than 0.2 MeV. Gross beta measurements may be used to measure emissions of specific radionuclides only (1) when it is known that the sample contains only a single radionuclide, and (2) measurements made using Method B-3 show reasonable agreement with the gross beta measurement. Gross beta measurements are applicable to mixtures of radionuclides only for the purposes and under the conditions described in section 3.7. APHA-602(4), ASTM-D-1890(11).
3.3.3 Method B-5, Liquid Scintillation Spectrometry.
Principle: An aliquot of a collected sample or the result of some other chemical separation or processing technique is added to a liquid scintillation “cocktail” which is viewed by photomultiplier tubes in a liquid scintillation spectrometer. The spectrometer is adjusted to establish a channel or “window” for the pulse energy appropriate to the nuclide of interest. The activity of the nuclide of interest is measured by the counting rate in the appropriate energy channel. Corrections are made for chemical yield where separations are made.
Applicability: This method is applicable to any beta-emitting nuclide when no other radionuclide is present in the sample or the separated sample provided that it can be incorporated in the scintillation cocktail. This method is also applicable for samples which contain more than one radionuclide but only when the energies of the beta particles are sufficiently separated so that they can be resolved by the spectrometer. This method is most applicable to the measurement of low-energy beta emitters such as tritium and carbon-14. APHA-609(6), EML-LV-539-17(19).
3.4 Gamma Emitting Radionuclides
3.4.1 Method G-1, High Resolution Gamma Spectrometry.
Principle: The sample is counted with a high resolution gamma detector, usually either a Ge(Li) or a high purity Ge detector, connected to a multichannel analyzer or computer. The gamma emitting radionuclides in the sample are measured from the gamma count rates in the energy regions characteristic of the individual radionuclide. Corrections are made for counts contributed by other radionuclides to the spectral regions of the radionuclides of interest. Radiochemical separations may be made prior to counting but are usually not necessary.
Applicability: This method is applicable to the measurement of any gamma emitting radionuclide with gamma energies greater than 20 keV. It can be applied to complex mixtures of radionuclides. The samples counted may be in the form of particulate filters, absorbers, liquids or gases. The method may also be applied to the analysis of gaseous gamma emitting radionuclides directly in an effluent stream by passing the stream through a chamber or cell containing the detector. ASTM-3649(9), IDO-12096(18).
3.4.2 Method G-2, Low Resolution Gamma Spectrometry.
Principle: The sample is counted with a low resolution gamma detector, a thallium activated sodium iodide crystal. The detector is coupled to a photomultiplier tube and connected to a multichannel analyzer. The gamma emitting radionuclides in the sample are measured from the gamma count rates in the energy regions characteristic of the individual radionuclides. Corrections are made for counts contributed by other radionuclides to the spectral regions of the radionuclides of interest. Radiochemical separation may be used prior to counting to obtain less complex gamma spectra if needed.
Applicability: This method is applicable to the measurement of gamma emitting radionuclides with energies greater than 100 keV. It can be applied only to relatively simple mixtures of gamma emitting radionuclides. The samples counted may be in the form of particulate filters, absorbers, liquids or gas. The method can be applied to the analysis of gaseous radionuclides directly in an effluent stream by passing the gas stream through a chamber or cell containing the detector. ASTM-D-2459(12), EMSL-LV-0539-17(19).
3.4.3 Method G-3, Single Channel Gamma Spectrometry.
Principle: The sample is counted with a thallium activated sodium iodide crystal. The detector is coupled to a photomultiplier tube connected to a single channel analyzer. The activity of a gamma emitting radionuclide is determined from the gamma counts in the energy range for which the counter is set.
Applicability: This method is applicable to the measurement of a single gamma emitting radionuclide. It is not applicable to mixtures of radionuclides. The samples counted may be in the form of particulate filters, absorbers, liquids or gas. The method can be applied to the analysis of gaseous radionuclides directly in an effluent stream by passing the gas stream through a chamber or cell containing the detector.
3.4.4 Method G-4, Gross Gamma Counting.
Principle: The sample is counted with a gamma detector usually a thallium activated sodium iodine crystal. The detector is coupled to a photomultiplier tube and gamma rays above a specific threshold energy level are counted.
Applicability: Gross gamma measurements may be used to measure emissions of specific radionuclides only when it is known that the sample contains a single radionuclide or the identity and isotopic ratio of the radionuclides in the effluent stream are well known. When gross gamma measurements are used to determine emissions of specific radionuclides periodic measurements using Methods G-1 or G-2 should be made to demonstrate that the gross gamma measurements provide reliable emission data. This method may be applied to analysis of gaseous radionuclides directly in an effluent stream by placing the detector directly in or adjacent to the effluent stream or passing an extracted sample of the effluent stream through a chamber or cell containing the detector.
3.5 Counting Methods. All of the above methods with the exception of Method A-5 involve counting the radiation emitted by the radionuclide. Counting methods applicable to the measurement of alpha, beta and gamma radiations are listed below. The equipment needed and the counting principles involved are described in detail in ASTM-3648(8).
3.5.1 Alpha Counting:
• Gas Flow Proportional Counters. The alpha particles cause ionization in the counting gas and the resulting electrical pulses are counted. These counters may be windowless or have very thin windows.
• Scintillation Counters. The alpha particles transfer energy to a scintillator resulting in a production of light photons which strike a photomultiplier tube converting the light photons to electrical pulses which are counted. The counters may involve the use of solid scintillation materials such as zinc sulfide or liquid scintillation solutions.
• Solid-State Counters. Semiconductor materials, such as silicon surface-barrier p-n junctions, act as solid ionization chambers. The alpha particles interact which the detector producing electron hole pairs. The charged pair is collected by an applied electrical field and the resulting electrical pulses are counted.
• Alpha Spectrometers. Semiconductor detectors used in conjunction with multichannel analyzers for energy discrimination.
3.5.2 Beta Counting:
• Ionization Chambers. These chambers contain the beta-emitting nuclide in gaseous form. The ionization current produced is measured.
• Geiger-Muller (GM) Counters-or Gas Flow Proportional Counters. The beta particles cause ionization in the counting gas and the resulting electrical pulses are counted. Proportional gas flow counters which are heavily shielded by lead or other metal, and provided with an anti-coincidence shield to reject cosmic rays, are called low background beta counters.
• Scintillation Counters. The beta particles transfer energy to a scintillator resulting in a production of light photons, which strike a photomultiplier tube converting the light photon to electrical pulses which are counted. This may involve the use of anthracene crystals, plastic scintillator, or liquid scintillation solutions with organic phosphors.
• Liquid Scintillation Spectrometers. Liquid scintillation counters which use two photomultiplier tubes in coincidence to reduce background counts. This counter may also electronically discriminate among pulses of a given range of energy.
3.5.3 Gamma Counting:
• Low-Resolution Gamma Spectrometers. The gamma rays interact with thallium activated sodium iodide or cesium iodide crystal resulting in the release of light photons which strike a photomultiplier tube converting the light pulses to electrical pulses proportional to the energy of the gamma ray. Multi-channel analyzers are used to separate and store the pulses according to the energy absorbed in the crystal.
• High-Resolution gamma Spectrometers. Gamma rays interact with a lithium-drifted (Ge(Li)) or high-purity germanium (HPGe) semiconductor detectors resulting in a production of electron-hole pairs. The charged pair is collected by an applied electrical field. A very stable low noise preamplifier amplifies the pulses of electrical charge resulting from the gamma photon interactions. Multichannel analyzers or computers are used to separate and store the pulses according to the energy absorbed in the crystal.
• Single Channel Analyzers. Thallium activated sodium iodide crystals used with a single window analyzer. Pulses from the photomultiplier tubes are separated in a single predetermined energy range.
3.5.4 Calibration of Counters. Counters are calibrated for specific radionuclide measurements using a standard of the radionuclide under either identical or very similar conditions as the sample to be counted. For gamma spectrometers a series of standards covering the energy range of interest may be used to construct a calibration curve relating gamma energy to counting efficiency.
In those cases where a standard is not available for a radionuclide, counters may be calibrated using a standard with energy characteristics as similar as possible to the radionuclide to be measured. For gross alpha and beta measurements of the unidentified mixtures of radionuclides, alpha counters are calibrated with a natural uranium standard and beta counters with a cesium-137 standard. The standard must contain the same weight and distribution of solids as the samples, and be mounted in an identical manner. If the samples contain variable amounts of solids, calibration curves relating weight of solids present to counting efficiency are prepared. Standards other than those prescribed may be used provided it can be shown that such standards are more applicable to the radionuclide mixture measured.
3.6 Radiochemical Methods for Selected Radionuclides. Methods for a selected list of radionuclides are listed in Table 1. The radionuclides listed are those which are most commonly used and which have the greatest potential for causing doses to members of the public. For radionuclides not listed in Table 1, methods based on any of the applicable “principles of measurement” described in section 3.1 through 3.4 may be used.
3.7 Applicability of Gross Alpha and Beta Measurements to Unidentified Mixtures of Radionuclides. Gross alpha and beta measurements may be used as a screening measurement as a part of an emission measurement program to identify the need to do specific radionuclide analyses or to confirm or verify that unexpected radionuclides are not being released in significant quantities.
Gross alpha (Method A-4) or gross beta (Methods B-2 or B-4) measurements may also be used for the purpose of comparing the measured concentrations in the effluent stream with the limiting “Concentration Levels for Environmental Compliance” in table 2 of appendix E. For unidentified mixtures, the measured concentration value shall be compared with the lowest environmental concentration limit for any radionuclide which is not known to be absent from the effluent stream.
Table 1 - List of Approved Methods for Specific Radionuclides
| Radionuclide | Approved methods of analysis |
|---|---|
| Am-241 | A-1, A-2, A-3, A-4 |
| Ar-41 | B-1, B-2, G-1, G-2, G-3, G-4 |
| Ba-140 | G-1, G-2, G-3, G-4 |
| Br-82 | G-1, G-2, G-3, G-4 |
| C-11 | B-1, B-2, G-1, G-2, G-3, G-4 |
| C-14 | B-5 |
| Ca-45 | B-3, B-4, B-5 |
| Ce-144 | G-1, G-2, G-3, G-4 |
| Cm-244 | A-1, A-2, A-3, A-4 |
| Co-60 | G-1, G-2, G-3, G-4 |
| Cr-51 | G-1, G-2, G-3, G-4 |
| Cs-134 | G-1, G-2, G-3, G-4 |
| Cs-137 | G-1, G-2, G-3, G-4 |
| Fe-55 | B-5, G-1 |
| Fe-59 | G-1, G-2, G-3, G-4 |
| Ga-67 | G-1, G-2, G-3, G-4 |
| H-3 (H2O) | B-5 |
| H-3 (gas) | B-1 |
| I-123 | G-1, G-2, G-3, G-4 |
| I-125 | G-1 |
| I-131 | G-1, G-2, G-3, G-4 |
| In-113m | G-1, G-2, G-3, G-4 |
| Ir-192 | G-1, G-2, G-3, G-4 |
| Kr-85 | B-1, B-2, B-5, G-1, G-2, G-3, G-4 |
| Kr-87 | B-1, B-2, G-1, G-2, G-3, G-4 |
| Kr-88 | B-1, B-2, G-1, G-2, G-3, G-4 |
| Mn-54 | G-1, G-2, G-3, G-4 |
| Mo-99 | G-1, G-2, G-3, G-4 |
| N-13 | B-1, B-2, G-1, G-2, G-3, G-4 |
| O-15 | B-1, B-2, G-1, G-2, G-3, G-4 |
| P-32 | B-3, B-4, B-5 |
| Pm-147 | B-3, B-4, B-5 |
| Po-210 | A-1, A-2, A-3, A-4 |
| Pu-238 | A-1, A-2, A-3, A-4 |
| Pu-239 | A-1, A-2, A-3, A-4 |
| Pu-240 | A-1, A-2, A-3, A-4 |
| Ra-226 | A-1, A-2, G-1, G-2 |
| S-35 | B-5 |
| Se-75 | G-1, G-2, G-3, G-4 |
| Sr-90 | B-3, B-4, B-5 |
| Tc-99 | B-3, B-4, B-5 |
| Te-201 | G-1, G-2, G-3, G-4 |
| Uranium (total alpha) | A-1, A-2, A-3, A-4 |
| Uranium (Isotopic) | A-1, A-3 |
| Uranium (Natural) | A-5 |
| Xe-133 | G-1 |
| Yb-169 | G-1, G-2, G-3, G-4 |
| Zn-65 | G-1, G-2, G-3, G-4 |
Each facility required to measure their radionuclide emissions shall conduct a quality assurance program in conjunction with the radionuclide emission measurements. This program shall assure that the emission measurements are representative, and are of known precision and accuracy and shall include administrative controls to assure prompt response when emission measurements indicate unexpectedly large emissions. The program shall consist of a system of policies, organizational responsibilities, written procedures, data quality specifications, audits, corrective actions and reports. This quality assurance program shall include the following program elements:
4.1 The organizational structure, functional responsibilities, levels of authority and lines of communications for all activities related to the emissions measurement program shall be identified and documented.
4.2 Administrative controls shall be prescribed to ensure prompt response in the event that emission levels increase due to unplanned operations.
4.3 The sample collection and analysis procedures used in measuring the emissions shall be described including where applicable:
4.3.1 Identification of sampling sites and number of sampling points, including the rationale for site selections.
4.3.2 A description of sampling probes and representativeness of the samples.
4.3.3 A description of any continuous monitoring system used to measure emissions, including the sensitivity of the system, calibration procedures and frequency of calibration.
4.3.4 A description of the sample collection systems for each radionuclide measured, including frequency of collection, calibration procedures and frequency of calibration.
4.3.5 A description of the laboratory analysis procedures used for each radionuclide measured, including frequency of analysis, calibration procedures and frequency of calibration.
4.3.6 A description of the sample flow rate measurement systems or procedures, including calibration procedures and frequency of calibration.
4.3.7 A description of the effluent flow rate measurement procedures, including frequency of measurements, calibration procedures and frequency of calibration.
4.4 The objectives of the quality assurance program shall be documented and shall state the required precision, accuracy and completeness of the emission measurement data including a description of the procedures used to assess these parameters. Accuracy is the degree of agreement of a measurement with a true or known value. Precision is a measure of the agreement among individual measurements of the same parameters under similar conditions. Completeness is a measure of the amount of valid data obtained compared to the amount expected under normal conditions.
4.5 A quality control program shall be established to evaluate and track the quality of the emissions measurement data against preset criteria. The program should include where applicable a system of replicates, spiked samples, split samples, blanks and control charts. The number and frequency of such quality control checks shall be identified.
4.6 A sample tracking system shall be established to provide for positive identification of samples and data through all phases of the sample collection, analysis and reporting system. Sample handling and preservation procedures shall be established to maintain the integrity of samples during collection, storage and analysis.
4.7 Regular maintenance, calibration and field checks shall be performed for each sampling system in use by satisfying the requirements found in Table 2: Maintenance, Calibration and Field Check Requirements.
Table 2 - Maintenance, Calibration and Field Check Requirements
| Sampling system components | Frequency of activity |
|---|---|
| Cleaning of thermal anemometer elements | As required by application. |
| Inspect pitot tubes for contaminant deposits | At least annually. |
| Inspect pitot tube systems for leaks | At least annually. |
| Inspect sharp-edged nozzles for damage | At least annually or after maintenance that could cause damage. |
| Check nozzles for alignment, presence of deposits, or other potentially degrading factors | Annually. |
| Check transport lines of HEPA-filtered applications to determine if cleaning is required | Annually. |
| Clean transport lines | Visible deposits for HEPA-filtered applications. Mean mass of deposited material exceeds 1g/m 2 for other applications. |
| Inspect or test the sample transport system for leaks | At least annually. |
| Check mass flow meters of sampling systems with a secondary or transfer standard | At least quarterly. |
| Inspect rotameters of sampling systems for presence of foreign matter | At the start of each sampling period. |
| Check response of stack flow rate systems | At least quarterly. |
| Calibration of flow meters of sampling systems | At least annually. |
| Calibration of effluent flow measurement devices | At least annually. |
| Calibration of timing devices | At least annually. |
4.8 Periodic internal and external audits shall be performed to monitor compliance with the quality assurance program. These audits shall be performed in accordance with written procedures and conducted by personnel who do not have responsibility for performing any of the operations being audited.
4.9 A corrective action program shall be established including criteria for when corrective action is needed, what corrective actions will be taken and who is responsible for taking the corrective action.
4.10 Periodic reports to responsible management shall be prepared on the performance of the emissions measurements program. These reports should include assessment of the quality of the data, results of audits and description of corrective actions.
4.11 The quality assurance program should be documented in a quality assurance project plan that should address each of the above requirements.
5. References(1) American National Standards Institute “Guide to Sampling Airborne Radioactive Materials in Nuclear Facilities”, ANSI-N13.1-1969, American National Standards Institute, New York, New York (1969).
(2) American Public Health Association, “Methods of Air Sampling”, 2nd Edition, Method 605, “Tentative Method of Analysis for Plutonium Content of Atmospheric Particulate Matter”. American Public Health Association, New York, NY (1977).
(3) Ibid, Method 601, “Tentative Method of Analysis for Gross Alpha Radioactivity Content of the Atmosphere”.
(4) Ibid, Method 602, “Tentative Method of the Analysis for Gross Beta Radioactivity Content of the Atmosphere”.
(5) Ibid, Method 608, “Tentative Method of Analysis for Strontium-90 Content of Atmospheric Particulate Matter”.
(6) Ibid, Method 609, “Tentative Method of Analysis for Tritium Content of the Atmosphere”.
(7) Ibid, Method 603, “Tentative Method of Analysis for Iodine-131 Content of the Atmosphere”.
(8) American Society for Testing and Materials, 1986 Annual Book ASTM Standards, Designation D-3648-78, “Standard Practices for the Measurement of Radioactivity”. American Society for Testing and Materials, Philadelphia, PA (1986).
(9) Ibid, Designation D-3649-85, “Standard Practice for High Resolution Gamma Spectrometry”.
(10) Ibid, Designation D-1943-81, “Standard Test Method for Alpha Particle Radioactivity of Water”.
(11) Ibid, Designation D-1890-81, “Standard Test Method for Beta Particle Radioactivity of Water”.
(12) Ibid, Designation D-2459-72, “Standard Test Method for Gamma Spectrometry of Water”.
(13) Ibid, Designation D-3972-82, “Standard Test Method for Isotopic Uranium in Water by Radiochemistry”.
(14) Ibid, Designation D-2907-83, “Standard Test Methods for Microquantities of Uranium in Water by Fluorometry”.
(15) Ibid, Designation E-318, “Standard Test Method for Uranium in Aqueous Solutions by Colorimetry”.
(16) Ibid, Designation D-3084-75, “Standard Practice for Alpha Spectrometry of Water”.
(17) Corley, J.P. and C.D. Corbit, “A Guide for Effluent Radiological Measurements at DOE Installations”, DOE/EP-0096, Pacific Northwest Laboratories, Richland, Washington (1983).
(18) Department of Energy, “RESL Analytical Chemistry Branch Procedures Manual”, IDO-12096, U.S. Department of Energy, Idaho Falls, Idaho (1982).
(19) Environmental Protection Agency, “Radiochemical Analytical Procedures for Analysis of Environmental Samples”, EMSL-LV-0539-17, U.S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory, Las Vegas, Nevada (1979).
(20) Environmental Protection Agency, “Radiochemistry Procedures Manual”, EPA 520/5-84-006, Eastern Environmental Radiation Facility, Montgomery, Alabama (1984).
(21) National Council on Radiation Protection and Measurements, NCRP Report No. 50, “Environmental Radiation Measurements”, National Council on Radiation Protection and Measurement, Bethesda, Maryland (1976).
(22) Ibid, Report No. 47, “Tritium Measurement Techniques”. (1976).
(23) Ibid, Report No. 58 “A Handbook of Radioactivity Measurement Procedures” (1985).
(24) Environmental Protection Agency, “Indoor Radon and Radon Decay Product Measurement Protocols”, EPA 520/1-89-009, U.S. Environmental Protection Agency, Washington, DC (1989).
Method 115 - Monitoring for Radon-222 EmissionsThis appendix describes the monitoring methods which must be used in determining the radon-222 emissions from underground uranium mines, uranium mill tailings piles, phosphogypsum stacks, and other piles of waste material emitting radon.
1. Radon-222 Emissions from Underground Uranium Mine Vents1.1 Sampling Frequency and Calculation of Emissions. Radon-222 emissions from underground uranium mine vents shall be determined using one of the following methods:
1.1.1 Continuous Measurement. These measurements shall be made and the emissions calculated as follows:
(a) The radon-222 concentration shall be continuously measured at each mine vent whenever the mine ventilation system is operational.
(b) Each mine vent exhaust flow rate shall be measured at least 4 times per year.
(c) A weekly radon-222 emission rate for the mine shall be calculated and recorded weekly as follows:
Aw= C1Q1T1 + C2Q2T2 + . . . CiQiTi Where: Aw = Total radon-222 emitted from the mine during week (Ci) Ci = Average radon-222 concentration in mine vent i(Ci/m 3) Qi = Volumetric flow rate from mine vent i(m 3/hr) Ti = Hours of mine ventilation system operation during week for mine vent i(hr)(d) The annual radon-222 emission rate is the sum of the weekly emission rates during a calendar year.
1.1.2 Periodic Measurement. This method is applicable only to mines that continuously operate their ventilation system except for extended shutdowns. Mines which start up and shut down their ventilation system frequently must use the continuous measurement method describe in Section 1.1.1 above. Emission rates determined using periodic measurements shall be measured and calculated as follows:
(a) The radon-222 shall be continuously measured at each mine vent for at least one week every three months.
(b) Each mine vent exhaust flow rate shall be measured at least once during each of the radon-222 measurement periods.
(c) A weekly radon-222 emission rate shall be calculated for each weekly period according to the method described in Section 1.1.1. In this calculation T = 168 hr.
(d) The annual radon-222 emission rate from the mine should be calculated as follows:
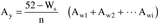 Where: Ay
= Annual radon-222 emission rate from the mine(Ci) Awi = Weekly
radon-222 emission rate during the measurement period i (Ci) n =
Number of weekly measurement periods per year Ws = Number of weeks
during the year that the mine ventilation system is shut down in
excess of 7 consecutive days, i.e. the sum of the number of weeks
each shut down exceeds 7 days
Where: Ay
= Annual radon-222 emission rate from the mine(Ci) Awi = Weekly
radon-222 emission rate during the measurement period i (Ci) n =
Number of weekly measurement periods per year Ws = Number of weeks
during the year that the mine ventilation system is shut down in
excess of 7 consecutive days, i.e. the sum of the number of weeks
each shut down exceeds 7 days
1.2 Test Methods and Procedures
Each underground mine required to test its emissions, unless an equivalent or alternative method has been approved by the Administrator, shall use the following test methods:
1.2.1 Test Method 1 of appendix A to part 60 shall be used to determine velocity traverses. The sampling point in the duct shall be either the centroid of the cross section or the point of average velocity.
1.2.2 Test Method 2 of appendix A to part 60 shall be used to determine velocity and volumetric flow rates.
1.2.3 Test Methods A-6 or A-7 of appendix B, Method 114 to part 61 shall be used for the analysis of radon-222. Use of Method A-7 requires prior approval of EPA based on conditions described in appendix B.
1.2.4 A quality assurance program shall be conducted in conformance with the programs described for Continuous Radon Monitors and Alpha Track Detectors in EPA 520/1-89-009. (2)
2. Radon-222 Emissions from Uranium Mill Tailings Piles2.1 Measurement and Calculation of Radon Flux from Uranium Mill Tailings Piles.
2.1.1 Frequency of Flux Measurement. A single set of radon flux measurements may be made, or if the owner or operator chooses, more frequent measurements may be made over a one year period. These measurements may involve quarterly, monthly or weekly intervals. All radon measurements shall be made as described in paragraphs 2.1.2 through 2.1.6 except that for measurements made over a one year period, the requirement of paragraph 2.1.4(c) shall not apply. The mean radon flux from the pile shall be the arithmetic mean of the mean radon flux for each measurement period. The weather conditions, moisture content of the tailings and area of the pile covered by water existing at the time of the measurement shall be chosen so as to provide measurements representative of the long term radon flux from the pile and shall be subject to EPA review and approval.
2.1.2 Distribution of Flux Measurements. The distribution and number of radon flux measurements required on a pile will depend on clearly defined areas of the pile (called regions) that can have significantly different radon fluxes due to surface conditions. The mean radon flux shall be determined for each individual region of the pile. Regions that shall be considered for operating mill tailings piles are:
(a) Water covered areas, (b) Water saturated areas (beaches), (c) Dry top surface areas, and (d) Sides, except where earthen material is used in dam construction.For mill tailings after disposal the pile shall be considered to consist of only one region.
2.1.3 Number of Flux Measurements. Radon flux measurements shall be made within each region on the pile, except for those areas covered with water. Measurements shall be made at regularly spaced locations across the surface of the region, realizing that surface roughness will prohibit measurements in some areas of a region. The minimum number of flux measurements considered necessary to determine a representative mean radon flux value for each type of region on an operating pile is:
(a) Water covered area - no measurements required as radon flux is assumed to be zero, (b) Water saturated beaches - 100 radon flux measurements, (c) Loose and dry top surface - 100 radon flux measurements, (d) Sides - 100 radon flux measurements, except where earthern material is used in dam construction.For a mill tailings pile after disposal which consists of only one region a minimum of 100 measurements are required.
2.1.4 Restrictions to Radon Flux Measurements. The following restrictions are placed on making radon flux measurements:
(a) Measurements shall not be initiated within 24 hours of a rainfall. (b) If a rainfall occurs during the 24 hour measurements period, the measurement is invalid if the seal around the lip of the collector has washed away or if the collector is surrounded by water. (c) Measurements shall not be performed if the ambient temperature is below 35 °F or if the ground is frozen.2.1.5 Areas of Pile Regions. The approximate area of each region of the pile shall be determined in units of square meters.
2.1.6 Radon Flux Measurement. Measuring radon flux involves the adsorption of radon on activated charcoal in a large-area collector. The radon collector is placed on the surface of the pile area to be measured and allowed to collect radon for a time period of 24 hours. The radon collected on the charcoal is measured by gamma-ray spectroscopy. The detailed measurement procedure provided in appendix A of EPA 520/5-85-0029(1) shall be used to measure the radon flux on uranium mill tailings, except the surface of the tailings shall not be penetrated by the lip of the radon collector as directed in the procedure, rather the collector shall be carefully positioned on a flat surface with soil or tailings used to seal the edge.
2.1.7 Calculations. The mean radon flux for each region of the pile and for the total pile shall be calculated and reported as follows:
(a) The individual radon flux calculations shall be made as provided in appendix A EPA 86 (1). The mean radon flux for each region of the pile shall be calculated by summing all individual flux measurements for the region and dividing by the total number of flux measurements for the region. (b) The mean radon flux for the total uranium mill tailings pile shall be calculated as follows.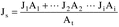 Where: Js = Mean flux for the total pile (pCi/m 2-s) Ji = Mean flux
measured in region i (pCi/m 2-s) Ai = Area of region i (m 2) At =
Total area of the pile (m 2)
Where: Js = Mean flux for the total pile (pCi/m 2-s) Ji = Mean flux
measured in region i (pCi/m 2-s) Ai = Area of region i (m 2) At =
Total area of the pile (m 2)
2.1.8 Reporting. The results of individual flux measurements, the approximate locations on the pile, and the mean radon flux for each region and the mean radon flux for the total stack shall be included in the emission test report. Any condition or unusual event that occurred during the measurements that could significantly affect the results should be reported.
3.0 Radon-222 Emissions from Phosphogypsum Stacks.
3.1 Measurement and Calculation of the Mean Radon Flux. Radon flux measurements shall be made on phosphogypsum stacks as described below:
3.1.1 Frequency of Measurements. A single set of radon flux measurements may be made after the phosphogypsum stack becomes inactive, or if the owner or operator chooses, more frequent measurements may be made over a one year period. These measurements may involve quarterly, monthly or weekly intervals. All radon measurements shall be made as described in paragraphs 3.1.2 through 3.1.6 except that for measurements made over a one year period, the requirement of paragraph 3.1.4(c) shall not apply. For measurements made over a one year period, the radon flux shall be the arithmetic mean of the mean radon flux for each measurement period.
3.1.2 Distribution and Number of Flux Measurements. The distribution and number of radon flux measurements required on a stack will depend on clearly defined areas of the stack (called regions) that can have significantly different radon fluxes due to surface conditions. The mean radon flux shall be determined for each individual region of the stack. Regions that shall be considered are:
(a) Water covered areas, (b) Water saturated areas (beaches), (c) Loose and dry top surface areas, (d) Hard-packed roadways, and (e) Sides.3.1.3 Number of Flux Measurements. Radon flux measurements shall be made within each region on the phosphogypsum stack, except for those areas covered with water. Measurements shall be made at regularly spaced locations across the surface of the region, realizing that surface roughness will prohibit measurements in some areas of a region. The minimum number of flux measurements considered necessary to determine a representative mean radon flux value for each type of region is:
(a) Water covered area - no measurements required as radon flux is assumed to be zero, (b) Water saturated beaches - 50 radon flux measurements, (c) Loose and dry top surface - 100 radon flux measurements, (d) Hard-packed roadways - 50 radon flux measurements, and (e) Sides - 100 radon flux measurements. A minimum of 300 measurements are required. A stack that has no water cover can be considered to consist of two regions, top and sides, and will require a minimum of only 200 measurements.3.1.4 Restrictions to Radon Flux Measurements. The following restrictions are placed on making radon flux measurements:
(a) Measurements shall not be initiated within 24 hours of a rainfall. (b) If a rainfall occurs during the 24 hour measurement period, the measurement is invalid if the seal around the lip of the collector has washed away or if the collector is surrounded by water. (c) Measurements shall not be performed if the ambient temperature is below 35 °F or if the ground is frozen.3.1.5 Areas of Stack Regions. The approximate area of each region of the stack shall be determined in units of square meters.
3.1.6 Radon Flux Measurements. Measuring radon flux involves the adsorption of radon on activated charcoal in a large-area collector. The radon collector is placed on the surface of the stack area to be measured and allowed to collect radon for a time period of 24 hours. The radon collected on the charcoal is measured by gamma-ray spectroscopy. The detailed measurement procedure provided in appendix A of EPA 520/5-85-0029(1) shall be used to measure the radon flux on phosphogypsum stacks, except the surface of the phosphogypsum shall not be penetrated by the lip of the radon collector as directed in the procedure, rather the collector shall be carefully positioned on a flat surface with soil or phosphogypsum used to seal the edge.
3.1.7 Calculations. The mean radon flux for each region of the phosphogypsum stack and for the total stack shall be calculated and reported as follows:
(a) The individual radon flux calculations shall be made as provided in appendix A EPA 86 (1). The mean radon flux for each region of the stack shall be calculated by summing all individual flux measurements for the region and dividing by the total number of flux measurements for the region. (b) The mean radon flux for the total phosphogypsum stack shall be calculated as follows.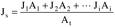 Where: Js
= Mean flux for the total stack (pCi/m 2-s) Ji = Mean flux measured
in region i (pCi/m 2-s) Ai = Area of region i (m 2) At = Total area
of the stack
Where: Js
= Mean flux for the total stack (pCi/m 2-s) Ji = Mean flux measured
in region i (pCi/m 2-s) Ai = Area of region i (m 2) At = Total area
of the stack
3.1.8 Reporting. The results of individual flux measurements, the approximate locations on the stack, and the mean radon flux for each region and the mean radon flux for the total stack shall be included in the emission test report. Any condition or unusual event that occurred during the measurements that could significantly affect the results should be reported.
4.0 Quality Assurance Procedures for Measuring Rn-222 Flux
A. Sampling ProceduresRecords of field activities and laboratory measurements shall be maintained. The following information shall be recorded for each charcoal canister measurement:
(a) Site (b) Name of pile (c) Sample location (d) Sample ID number (e) Date and time on (f) Date and time off (g) Observations of meteorological conditions and commentsRecords shall include all applicable information associated with determining the sample measurement, calculations, observations, and comments.
B. Sample CustodyCustodial control of all charcoal samples exposed in the field shall be maintained in accordance with EPA chain-of-custody field procedures. A control record shall document all custody changes that occur between the field and laboratory personnel.
C. Calibration Procedures and FrequencyThe radioactivity of two standard charcoal sources, each containing a carefully determined quantity of radium-226 uniformly distributed through 180g of activated charcoal, shall be measured. An efficiency factor is computed by dividing the average measured radioactivity of the two standard charcoal sources, minus the background, in cpm by the known radioactivity of the charcoal sources in dpm. The same two standard charcoal sources shall be counted at the beginning and at the end of each day's counting as a check of the radioactivity counting equipment. A background count using unexposed charcoal should also be made at the beginning and at the end of each counting day to check for inadvertent contamination of the detector or other changes affecting the background. The unexposed charcoal comprising the blank is changed with each new batch of charcoal used.
D. Internal Quality Control Checks and FrequencyThe charcoal from every tenth exposed canister shall be recounted. Five percent of the samples analyzed shall be either blanks (charcoal having no radioactivity added) or samples spiked with known quantities of radium-226.
E. Data Precision, Accuracy, and CompletenessThe precision, accuracy, and completeness of measurements and analyses shall be within the following limits for samples measuring greater than 1.0 pCi/m 2−s.
(a) Precision: 10%
(b) Accuracy: ±10%
(c) Completeness: at least 85% of the measurements must yield useable results.
5.0 References(1) Hartley, J.N. and Freeman, H.D., “Radon Flux Measurements on Gardinier and Royster phosphogypsum Piles Near Tampa and Mulberry, Florida,” U.S. Environmental Protection Agency Report, EPA 520/5-85-029, January 1986.
(2) Environmental Protection Agency, “Indoor Radon and Radon Decay Product Measurement Protocols”, EPA 520/1-89-009, U.S. Environmental Protection Agency, Washington, DC. (1989).
[38 FR 8826, Apr. 6, 1973] Editorial Note:For Federal Register citations affecting appendix B, see the List of CFR Sections Affected, which appears in the Finding Aids section of the printed volume and at www.govinfo.gov.Appendix C to Part 61 - Quality Assurance Procedures
40:10.0.1.1.1.29.1.21.6 : Appendix C
Appendix C to Part 61 - Quality Assurance Procedures Procedure 1 - Determination of Adequate Chromatographic Peak ResolutionIn this method of dealing with resolution, the extent to which one chromatographic peak overlaps another is determined.
For convenience, consider the range of the elution curve of each compound as running from −2σ to + 2σ. This range is used in other resolution criteria, and it contains 95.45 percent of the area of a normal curve. If two peaks are separated by a known distance, b, one can determine the fraction of the area of one curve that lies within the range of the other. The extent to which the elution curve of a contaminant compound overlaps the curve of a compound that is under analysis is found by integrating the contaminant curve over the limits b−2σs to b + 2σs, where σs is the standard deviation of the sample curve.
This calculation can be simplified in several ways. Overlap can be determined for curves of unit area; then actual areas can be introduced. Desired integration can be resolved into two integrals of the normal distribution function for which there are convenient calculation programs and tables. An example would be Program 15 in Texas Instruments Program Manual ST1, 1975, Texas Instruments, Inc., Dallas, Texas 75222.

In judging the suitability of alternate GC columns or the effects of altering chromatographic conditions, one can employ the area overlap as the resolution parameter with a specific maximum permissible value.
The use of Gaussian functions to describe chromatographic elution curves is widespread. However, some elution curves are highly asymmetric. In cases where the sample peak is followed by a contaminant that has a leading edge that rises sharply but the curve then tails off, it may be possible to define an effective width for tc as “twice the distance from the leading edge to a perpendicular line through the maxim of the contaminant curve, measured along a perpendicular bisection of that line.”
Procedure 2 - Procedure for Field Auditing GC AnalysisResponsibilities of audit supervisor and analyst at the source sampling site include the following:
A. The audit supervisor verifies that audit cylinders are stored in a safe location both before and after the audit to prevent vandalism.
B. At the beginning and conclusion of the audit, the analyst records each cylinder number and pressure. An audit cylinder is never analyzed when the pressure drops below 200 psi.
C. During the audit, the analyst performs a minimum of two consecutive analyses of each audit cylinder gas. The audit must be conducted to coincide with the analysis of source test samples, normally immediately after GC calibration and prior to sample analyses.
D. At the end of audit analyses, the audit supervisor requests the calculated concentrations from the analyst and compares the results with the actual audit concentrations. If each measured concentration agrees with the respective actual concentration within ±10 percent, he directs the analyst to begin analyzing source samples. Audit supervisor judgment and/or supervisory policy determine action when agreement is not within ±10 percent. When a consistent bias in excess of 10 percent is found, it may be possible to proceed with the sample analysis, with a corrective factor to be applied to the results at a later time. However, every attempt should be made to locate the cause of the discrepancy, as it may be misleading. The audit supervisor records each cylinder number, cylinder pressure (at the end of the audit), and all calculated concentrations. The individual being audited must not under any circumstance be told actual audit concentrations until calculated concentrations have been submitted to the audit supervisor.
Field Audit ReportPart A - To be filled out by organization supplying audit cylinders.
1. Organization supplying audit sample(s) and shipping address
2. Audit supervisor, organization, and phone number
3. Shipping instructions: Name, Address, Attention
4. Guaranteed arrival date for cylinders 5. Planned shipping date for cylinders6. Details on audit cylinders from last analysis
| Low conc. | High conc. | |
|---|---|---|
| a. Date of last analysis | ||
| b. Cylinder number | ||
| c. Cylinder pressure, psi | ||
| d. Audit gas(es)/balance gas | ||
| e. Audit gas(es), ppm | ||
| f. Cylinder construction |
Part B - To be filled out by audit supervisor.
1. Process sampled 2. Audit location 3. Name of individual audit 4. Audit date5. Audit results:
| Low conc. cylinder | High conc. cylinder | |
|---|---|---|
| a. Cylinder number | ||
| b. Cylinder pressure before audit, psi | ||
| c. Cylinder pressure after audit, psi | ||
| d. Measured concentration, ppm Injection #1* Injection #2* Average | ||
| e. Actual audit concentration, ppm (Part A, 6e) | ||
| f. Audit accuracy: 1 | ||
| Low Conc. Cylinder | ||
| High Conc. Cylinder | ||
| Percent 1 accuracy= | ||
| Measured Conc. − Actual Conc. | ||
| ________________ × 100 | ||
| Actual Conc. | ||
| g. Problems detected (if any) |
1 Results of two consecutive injections that meet the sample analysis criteria of the test method.
Appendix D to Part 61 - Methods for Estimating Radionuclide Emissions
40:10.0.1.1.1.29.1.21.7 : Appendix D
Appendix D to Part 61 - Methods for Estimating Radionuclide Emissions 1. Purpose and BackgroundFacility owners or operators may estimate radionuclide emissions to the atmosphere for dose calculations instead of measuring emissions. Particulate emissions from mill tailings piles should be estimated using the procedures listed in reference re #2. All other emissions may be estimated by using the “Procedures” listed below, or using the method described in reference #1.
2. ProcedureTo estimate emissions to the atmosphere:
(a) Determine the amount (in curies) used at facilities for the period under consideration. Radioactive materials in sealed packages that remain unopened, and have not leaked during the assessment period should not be included in the calculation.
(b) Multiply the amount used by the following factors which depend on the physical state of the radionuclide. They are:
(i) 1 for gases;
(ii) 10−3 for liquids or particulate solids; and
(iii) 10−6 for solids.
If any nuclide is heated to a temperature of 100 degrees Celsius or more, boils at a temperature of 100 degrees Celsius or less, or is intentionally dispersed into the environment, it must be considered to be a gas.
(c) If a control device is installed between the place of use and the point of release, multiply emissions from (b) by an adjustment factor. These are presented in Table 1.
Table 1 - Adjustment to Emission Factors for Effluent Controls
| Controls | Types of radionuclides controlled | Adjustment factor to emissions | Comments and conditions |
|---|---|---|---|
| HEPA filters | Particulates | 0.01 | Not applicable to gaseous radionuclides; periodic testing is prudent to ensure high removal efficiency. |
| Fabric filter | Particulates | 0.1 | Monitoring would be prudent to guard against tears in filter. |
| Sintered metal | Particulates | 1 | Insufficient data to make recommendation. |
| Activated carbon filters | Iodine gas | 0.1 | Efficiency is time dependent; monitoring is necessary to ensure effectiveness. |
| Douglas bags: Held one week or longer for decay | Xenon | 0.5/wk | Based on xenon half-life of 5.3 days; |
| Douglas bags: Released within one week | Xenon | 1 | Provides no reduction of exposure to general public. |
| Venturi scrubbers | Particulates Gases |
0.05 1 |
Although venturis may remove gases, variability in gaseous removal efficiency dictates adjustment factor for particulates only. |
| Packed bed scrubbers | Gases | 0.1 | Not applicable to particulates. |
| Electrostatic precipitators | Particulates | 0.05 | Not applicable for gaseous radionuclides |
| Xenon traps | Xenon | 0.1 | Efficiency is time dependent; monitoring is necessary to ensure effectiveness. |
| Fume hoods | All | 1 | Provides no reduction to general public exposures. |
| Vent stacks | All | 1 | Generally provides no reduction of exposure to general public. |
(1) Environmental Protection Agency, “A Guide for Determining Compliance with the Clean Air Act Standards for Radionuclides Emissions from NRC-Licensed and Non-DOE Federal Facilities”, EPA 520/1-89-002, January 1989.
(2) Nuclear Regulatory Commission, “Methods for Estimating Radioactive and Toxic Airborne Source Terms for Uranium Milling Operations”, U.S. Nuclear Regulatory Commission Regulatory Guide 3.59, March 1987.
[54 FR 51711, Dec. 15, 1989]Appendix E to Part 61 - Compliance Procedures Methods for Determining Compliance With Subpart I
40:10.0.1.1.1.29.1.21.8 : Appendix E
Appendix E to Part 61 - Compliance Procedures Methods for Determining Compliance With Subpart I 1. Purpose and BackgroundThis Appendix provides simplified procedures to reduce the burden on Nuclear Regulatory Commission (NRC) licensees, and non-Department of Energy Federal facilities in determining compliance with 40 CFR part 61, subpart I. The procedures consist of a series of increasingly more stringent steps, depending on the facility's potential to exceed the standard.
First, a facility can be found in compliance if the quantity of radioactive material possessed during the year is less than that listed in a table of annual possession quantities. A facility will also be in compliance if the average annual radionuclide emission concentration is less than that listed in a table of air concentration levels. If the facility is not in compliance by these tables, it can establish compliance by estimating a dose using screening procedure developed by the National Council on Radiation Protection and Measurements with a radiological source term derived using EPA approved emission factors. These procedures are described in a “Guide for Determining Compliance with the Clean Air Act Standards for Radionuclide Emissions From NRC-Licensed and Non-DOE Federal Facilities.”
A user-friendly computer program called COMPLY has been developed to reduce the burden on the regulated community. The Agency has also prepared a “User's Guide for the COMPLY Code” to assist the regulated community in using the code, and in handling more complex situations such as multiple release points. The basis for these compliance procedures are provided in “Background Information Document: Procedures Approved for Demonstrating Compliance with 40 CFR Part 61, Subpart I”. The compliance model is the highest level in the COMPLY computer code and provides for the most realistic assessment of dose by allowing the use of site-specific information.
2. Table of Annual Possession Quantity(a) Table 1 may be used for determining if facilities are in compliance with the standard. The possession table can only be used if the following conditions are met:
(i) No person lives within 10 meters of any release point; and
(ii) No milk, meat, or vegetables are produced within 100 meters of any release point.
(b) Procedures described in Reference (1) shall be used to determine compliance or exemption from reporting by use of Table 2.
Table 1 - Annual Possession Quantities for Environmental Compliance
[Annual Possession Quantities (Ci/yr)]
| Radionuclide | Gaseous form* | Liquid/powder forms | Solid form* |
|---|---|---|---|
| Ac-225 | 9.6E−05 | 9.6E−02 | 9.6E + 01 |
| Ac-227 | 1.6E−07 | 1.6E−04 | 1.6E−01 |
| Ac-228 | 3.4E−03 | 3.4E + 00 | 3.4E + 03 |
| Ag-106 | 1.6E + 00 | 1.6E + 03 | 1.6E + 06 |
| Ag-106m | 2.6E−03 | 2.6E + 00 | 2.6E + 03 |
| Ag-108m | 6.5E−06 | 6.5E−03 | 6.5E + 00 |
| Ag-110m | 9.4E−05 | 9.4E−02 | 9.4E + 01 |
| Ag-111 | 6.7E−02 | 6.7E + 01 | 6.7E + 04 |
| Al-26 | 4.0E−06 | 4.0E−03 | 4.0E + 00 |
| Am-241 | 2.3E−06 | 2.3E−03 | 2.3E + 00 |
| Am-242 | 1.8E−02 | 1.8E + 01 | 1.8E + 04 |
| Am-242m | 2.5E−06 | 2.5E−03 | 2.5E + 00 |
| Am-243 | 2.3E−06 | 2.3E−03 | 2.3E + 00 |
| Am-244 | 4.6E−02 | 4.6E + 01 | 4.6E + 04 |
| Am-245 | 7.0E + 00 | 7.0E + 03 | 7.0E + 06 |
| Am-246 | 9.8E−01 | 9.8E + 02 | 9.8E + 05 |
| Ar-37 | 1.4E + 06 | ||
| Ar-41 | 1.4E + 00 | ||
| As-72 | 2.9E−02 | 2.9E + 01 | 2.9E + 04 |
| As-73 | 6.0E−02 | 6.0E + 01 | 6.0E + 04 |
| As-74 | 4.3E−03 | 4.3E + 00 | 4.3E + 03 |
| As-76 | 8.8E−02 | 8.8E + 01 | 8.8E + 04 |
| As-77 | 7.9E−01 | 7.9E + 02 | 7.9E + 05 |
| At-211 | 1.0E−02 | 1.0E + 01 | 1.0E + 04 |
| Au-193 | 4.2E−01 | 4.2E + 02 | 4.2E + 05 |
| Au-194 | 3.5E−02 | 3.5E + 01 | 3.5E + 04 |
| Au-195 | 3.3E−03 | 3.3E + 00 | 3.3E + 03 |
| Au-198 | 4.6E−02 | 4.6E + 01 | 4.6E + 04 |
| Au-199 | 1.5E−01 | 1.5E + 02 | 1.5E + 05 |
| Ba-131 | 1.0E−02 | 1.0E + 01 | 1.0E + 04 |
| Ba-133 | 4.9E−05 | 4.9E−02 | 4.9E + 01 |
| Ba-133m | 9.3E−02 | 9.3E + 01 | 9.3E + 04 |
| Ba-135m | 5.8E−01 | 5.8E + 02 | 5.8E + 05 |
| Ba-139 | 4.7E + 00 | 4.7E + 03 | 4.7E + 06 |
| Ba-140 | 2.1E−03 | 2.1E + 00 | 2.1E + 03 |
| Ba-141 | 1.3E + 00 | 1.3E + 03 | 1.3E + 06 |
| Ba-142 | 1.1E + 00 | 1.1E + 03 | 1.1E + 06 |
| Be-7 | 2.3E−02 | 2.3E + 01 | 2.3E + 04 |
| Be-10 | 3.0E−03 | 3.0E + 00 | 3.0E + 03 |
| Bi-206 | 3.1E−03 | 3.1E + 00 | 3.1E + 03 |
| Bi-207 | 8.4E−06 | 8.4E−03 | 8.4E + 00 |
| Bi-210 | 4.2E−03 | 4.2E + 00 | 4.2E + 03 |
| Bi-212 | 4.7E−02 | 4.7E + 01 | 4.7E + 04 |
| Bi-213 | 6.0E−02 | 6.0E + 01 | 6.0E + 04 |
| Bi-214 | 1.4E−01 | 1.4E + 02 | 1.4E + 05 |
| Bk-249 | 7.0E−04 | 7.0E−01 | 7.0E + 02 |
| Bk-250 | 1.0E−01 | 1.0E + 02 | 1.0E + 05 |
| Br-77 | 7.5E−02 | 7.5E + 01 | 7.5E + 04 |
| Br-80 | 1.2E + 01 | 1.2E + 04 | 1.2E + 07 |
| Br-80m | 1.5E + 00 | 1.5E + 03 | 1.5E + 06 |
| Br-82 | 1.6E−02 | 1.6E + 01 | 1.6E + 04 |
| Br-83 | 9.9E + 00 | 9.9E + 03 | 9.9E + 06 |
| Br-84 | 5.6E−01 | 5.6E + 02 | 5.6E + 05 |
| C-11 | 1.3E + 00 | 1.3E + 03 | 1.3E + 06 |
| C-14 | 2.9E−01 | 2.9E + 02 | 2.9E + 05 |
| Ca-41 | 2.7E−02 | 2.7E + 01 | 2.7E + 04 |
| Ca-45 | 5.8E−02 | 5.8E + 01 | 5.8E + 04 |
| Ca-47 | 1.1E−02 | 1.1E + 01 | 1.1E + 04 |
| Cd-109 | 5.0E−03 | 5.0E + 00 | 5.0E + 03 |
| Cd-113 | 3.3E−04 | 3.3E−01 | 3.3E + 02 |
| Cd-113m | 4.4E−04 | 4.4E−01 | 4.4E + 02 |
| Cd-115 | 5.4E−02 | 5.4E + 01 | 5.4E + 04 |
| Cd-115m | 1.0E−02 | 1.0E + 01 | 1.0E + 04 |
| Cd-117 | 5.6E−02 | 5.6E + 01 | 5.6E + 04 |
| Cd-117m | 1.3E−01 | 1.3E + 02 | 1.3E + 05 |
| Ce-139 | 2.6E−03 | 2.6E + 00 | 2.6E + 03 |
| Ce-141 | 1.8E−02 | 1.8E + 01 | 1.8E + 04 |
| Ce-143 | 1.0E−01 | 1.0E + 02 | 1.0E + 05 |
| Ce-144 | 1.7E−03 | 1.7E + 00 | 1.7E + 03 |
| Cf-248 | 2.0E−05 | 2.0E−02 | 2.0E + 01 |
| Cf-249 | 1.7E−06 | 1.7E−03 | 1.7E + 00 |
| Cf-250 | 4.0E−06 | 4.0E−03 | 4.0E + 00 |
| Cf-251 | 1.7E−06 | 1.7E−03 | 1.7E + 00 |
| Cf-252 | 6.4E−06 | 6.4E−03 | 6.4E + 00 |
| Cf-253 | 3.3E−04 | 3.3E−01 | 3.3E + 02 |
| Cf-254 | 3.6E−06 | 3.6E−03 | 3.6E + 00 |
| Cl-36 | 1.9E−04 | 1.9E−01 | 1.9E + 02 |
| Cl-38 | 6.5E−01 | 6.5E + 02 | 6.5E + 05 |
| Cm-242 | 6.0E−05 | 6.0E−02 | 6.0E + 01 |
| Cm-243 | 3.3E−06 | 3.3E−03 | 3.3E + 00 |
| Cm-244 | 4.2E−06 | 4.2E−03 | 4.2E + 00 |
| Cm-245 | 2.3E−06 | 2.3E−03 | 2.3E + 00 |
| Cm-246 | 2.3E−06 | 2.3E−03 | 2.3E + 00 |
| Cm-247 | 2.3E−06 | 2.3E−03 | 2.3E + 00 |
| Cm-248 | 6.4E−07 | 6.4E−04 | 6.4E−01 |
| Cm-249 | 4.6E + 00 | 4.6E + 03 | 4.6E + 06 |
| Cm-250 | 1.1E−07 | 1.1E−04 | 1.1E−01 |
| Co-56 | 2.4E−04 | 2.4E−01 | 2.4E + 02 |
| Co-57 | 1.6E−03 | 1.6E + 00 | 1.6E + 03 |
| Co-58 | 9.0E−04 | 9.0E−01 | 9.0E + 02 |
| Co-58m | 1.7E−01 | 1.7E + 02 | 1.7E + 05 |
| Co-60 | 1.6E−05 | 1.6E−02 | 1.6E + 01 |
| Co-60m | 4.0E + 00 | 4.0E + 03 | 4.0E + 06 |
| Co-61 | 3.8E + 00 | 3.8E + 03 | 3.8E + 06 |
| Cr-49 | 9.0E−01 | 9.0E + 02 | 9.0E + 05 |
| Cr-51 | 6.3E−02 | 6.3E + 01 | 6.3E + 04 |
| Cs-129 | 1.5E−01 | 1.5E + 02 | 1.5E + 05 |
| Cs-131 | 2.8E−01 | 2.8E + 02 | 2.8E + 05 |
| Cs-132 | 1.3E−02 | 1.3E + 01 | 1.3E + 04 |
| Cs-134 | 5.2E−05 | 5.2E−02 | 5.2E + 01 |
| Cs-134m | 3.2E−01 | 3.2E + 02 | 3.2E + 05 |
| Cs-135 | 2.4E−02 | 2.4E + 01 | 2.4E + 04 |
| Cs-136 | 2.1E−03 | 2.1E + 00 | 2.1E + 03 |
| Cs-137 | 2.3E−05 | 2.3E−02 | 2.3E + 01 |
| Cs-138 | 4.4E−01 | 4.4E + 02 | 4.4E + 05 |
| Cu-61 | 4.0E−01 | 4.0E + 02 | 4.0E + 05 |
| Cu-64 | 5.2E−01 | 5.2E + 02 | 5.2E + 05 |
| Cu-67 | 1.5E−01 | 1.5E + 02 | 1.5E + 05 |
| Dy-157 | 4.4E−01 | 4.4E + 02 | 4.4E + 05 |
| Dy-165 | 5.6E + 00 | 5.6E + 03 | 5.6E + 06 |
| Dy-166 | 8.1E−02 | 8.1E + 01 | 8.1E + 04 |
| Er-169 | 4.0E−01 | 4.0E + 02 | 4.0E + 05 |
| Er-171 | 3.6E−01 | 3.6E + 02 | 3.6E + 05 |
| Es-253 | 2.6E−04 | 2.6E−01 | 2.6E + 02 |
| Es-254 | 2.3E−05 | 2.3E−02 | 2.3E + 01 |
| Es-254m | 1.8E−03 | 1.8E + 00 | 1.8E + 03 |
| Eu-152 | 1.6E−05 | 1.6E−02 | 1.6E + 01 |
| Eu-152m | 3.5E−01 | 3.5E + 02 | 3.5E + 05 |
| Eu-154 | 2.0E−05 | 2.0E−02 | 2.0E + 01 |
| Eu-155 | 5.2E−04 | 5.2E−01 | 5.2E + 02 |
| Eu-156 | 3.2E−03 | 3.2E + 00 | 3.2E + 03 |
| F-18 | 5.6E−01 | 5.6E + 02 | 5.6E + 05 |
| Fe-52 | 4.9E−02 | 4.9E + 01 | 4.9E + 04 |
| Fe-55 | 1.4E−01 | 1.4E + 02 | 1.4E + 05 |
| Fe-59 | 1.3E−03 | 1.3E + 00 | 1.3E + 03 |
| Fm-254 | 1.8E−02 | 1.8E + 01 | 1.8E + 04 |
| Fm-255 | 4.0E−03 | 4.0E + 00 | 4.0E + 03 |
| Fr-223 | 1.4E−01 | 1.4E + 02 | 1.4E + 05 |
| Ga-66 | 5.6E−02 | 5.6E + 01 | 5.6E + 04 |
| Ga-67 | 1.1E−01 | 1.1E + 02 | 1.1E + 05 |
| Ga-68 | 7.6E−01 | 7.6E + 02 | 7.6E + 05 |
| Ga-72 | 3.6E−02 | 3.6E + 01 | 3.6E + 04 |
| Gd-152 | 4.4E−06 | 4.4E−03 | 4.4E + 00 |
| Gd-153 | 2.0E−03 | 2.0E + 00 | 2.0E + 03 |
| Gd-159 | 6.8E−01 | 6.8E + 02 | 6.8E + 05 |
| Ge-68 | 2.3E−04 | 2.3E−01 | 2.3E + 02 |
| Ge-71 | 2.6E + 00 | 2.6E + 03 | 2.6E + 06 |
| Ge-77 | 1.0E−01 | 1.0E + 02 | 1.0E + 05 |
| H-3 | 1.5E + 01 | 1.5E + 04 | 1.5E + 07 |
| Hf-181 | 2.5E−03 | 2.5E + 00 | 2.5E + 03 |
| Hg-193m | 9.5E−02 | 9.5E + 01 | 9.5E + 04 |
| Hg-197 | 2.4E−01 | 2.4E + 02 | 2.4E + 05 |
| Hg-197m | 2.5E−01 | 2.5E + 02 | 2.5E + 05 |
| Hg-203 | 5.2E−03 | 5.2E + 00 | 5.2E + 03 |
| Ho-166 | 2.8E−01 | 2.8E + 02 | 2.8E + 05 |
| Ho-166m | 6.0E−06 | 6.0E−03 | 6.0E + 00 |
| I-123 | 4.9E−01 | 4.9E + 02 | 4.9E + 05 |
| I-124 | 9.3E−03 | 9.3E + 00 | 9.3E + 03 |
| I-125 | 6.2E−03 | 6.2E + 00 | 6.2E + 03 |
| I-126 | 3.7E−03 | 3.7E + 00 | 3.7E + 03 |
| I-128 | 9.3E + 00 | 9.3E + 03 | 9.3E + 06 |
| I-129 | 2.6E−04 | 2.6E−01 | 2.6E + 02 |
| I-130 | 4.6E−02 | 4.6E + 01 | 4.6E + 04 |
| I-131 | 6.7E−03 | 6.7E + 00 | 6.7E + 03 |
| I-132 | 2.0E−01 | 2.0E + 02 | 2.0E + 05 |
| I-133 | 6.7E−02 | 6.7E + 01 | 6.7E + 04 |
| I-134 | 3.2E−01 | 3.2E + 02 | 3.2E + 05 |
| I-135 | 1.2E−01 | 1.2E + 02 | 1.2E + 05 |
| In-111 | 4.9E−02 | 4.9E + 01 | 4.9E + 04 |
| In-113m | 2.1E + 00 | 2.1E + 03 | 2.1E + 06 |
| In-114m | 4.9E−03 | 4.9E + 00 | 4.9E + 03 |
| In-115 | 2.7E−04 | 2.7E−01 | 2.7E + 02 |
| In-115m | 1.4E + 00 | 1.4E + 03 | 1.4E + 06 |
| In-116m | 3.5E−01 | 3.5E + 02 | 3.5E + 05 |
| In-117 | 1.3E + 00 | 1.3E + 03 | 1.3E + 06 |
| In-117m | 7.6E−02 | 7.6E + 01 | 7.6E + 04 |
| Ir-190 | 3.5E−03 | 3.5E + 00 | 3.5E + 03 |
| Ir-192 | 9.7E−04 | 9.7E−01 | 9.7E + 02 |
| Ir-194 | 2.5E−01 | 2.5E + 02 | 2.5E + 05 |
| Ir-194m | 1.5E−04 | 1.5E−01 | 1.5E + 02 |
| K-40 | 6.8E−05 | 6.8E−02 | 6.8E + 01 |
| K-42 | 2.9E−01 | 2.9E + 02 | 2.9E + 05 |
| K-43 | 6.0E−02 | 6.0E + 01 | 6.0E + 04 |
| K-44 | 4.9E−01 | 4.9E + 02 | 4.9E + 05 |
| Kr-79 | 7.0E + 00 | ||
| Kr-81 | 1.8E + 02 | ||
| Kr-83m | 2.0E + 04 | ||
| Kr-85 | 8.4E + 02 | ||
| Kr-85m | 1.1E + 01 | ||
| Kr-87 | 2.0E + 00 | ||
| Kr-88 | 4.2E−01 | ||
| La-140 | 1.6E−02 | 1.6E + 01 | 1.6E + 04 |
| La-141 | 1.1E + 00 | 1.1E + 03 | 1.1E + 06 |
| La-142 | 2.3E−01 | 2.3E + 02 | 2.3E + 05 |
| Lu-177 | 1.4E−01 | 1.4E + 02 | 1.4E + 05 |
| Lu-177m | 3.5E−04 | 3.5E−01 | 3.5E + 02 |
| Mg-28 | 2.1E−02 | 2.1E + 01 | 2.1E + 04 |
| Mn-52 | 3.5E−03 | 3.5E + 00 | 3.5E + 03 |
| Mn-52m | 5.2E−01 | 5.2E + 02 | 5.2E + 05 |
| Mn-53 | 5.7E−02 | 5.7E + 01 | 5.7E + 04 |
| Mn-54 | 2.5E−04 | 2.5E−01 | 2.5E + 02 |
| Mn-56 | 2.5E−01 | 2.5E + 02 | 2.5E + 05 |
| Mo-93 | 1.5E−03 | 1.5E + 00 | 1.5E + 03 |
| Mo-99** | 5.7E−02 | 5.7E + 01 | 5.7E + 04 |
| Mo-101 | 8.4E−01 | 8.4E + 02 | 8.4E + 05 |
| Na-22 | 3.2E−05 | 3.2E−02 | 3.2E + 01 |
| Na-24 | 2.6E−02 | 2.6E + 01 | 2.6E + 04 |
| Nb-90 | 2.5E−02 | 2.5E + 01 | 2.5E + 04 |
| Nb-93m | 1.2E−02 | 1.2E + 01 | 1.2E + 04 |
| Nb-94 | 6.0E−06 | 6.0E−03 | 6.0E + 00 |
| Nb-95 | 2.3E−03 | 2.3E + 00 | 2.3E + 03 |
| Nb-95m | 2.0E−02 | 2.0E + 01 | 2.0E + 04 |
| Nb-96 | 2.5E−02 | 2.5E + 01 | 2.5E + 04 |
| Nb-97 | 1.0E + 00 | 1.0E + 03 | 1.0E + 06 |
| Nd-147 | 3.0E−02 | 3.0E + 01 | 3.0E + 04 |
| Nd-149 | 1.1E + 00 | 1.1E + 03 | 1.1E + 06 |
| Ni-56 | 2.0E−03 | 2.0E + 00 | 2.0E + 03 |
| Ni-57 | 2.1E−02 | 2.1E + 01 | 2.1E + 04 |
| Ni-59 | 2.2E−02 | 2.2E + 01 | 2.2E + 04 |
| Ni-63 | 1.4E−01 | 1.4E + 02 | 1.4E + 05 |
| Ni-65 | 7.0E−01 | 7.0E + 02 | 7.0E + 05 |
| Np-235 | 3.0E−02 | 3.0E + 01 | 3.0E + 04 |
| Np-237 | 1.8E−06 | 1.8E−03 | 1.8E + 00 |
| Np-238 | 1.9E−02 | 1.9E + 01 | 1.9E + 04 |
| Np-239 | 1.0E−01 | 1.0E + 02 | 1.0E + 05 |
| Np-240 | 6.5E−01 | 6.5E + 02 | 6.5E + 05 |
| Np-240m | 4.7E + 00 | 4.7E + 03 | 4.7E + 06 |
| Os-185 | 9.2E−04 | 9.2E−01 | 9.2E + 02 |
| Os-191m | 9.0E−01 | 9.0E + 02 | 9.0E + 05 |
| Os-191 | 3.8E−02 | 3.8E + 01 | 3.8E + 04 |
| Os-193 | 2.9E−01 | 2.9E + 02 | 2.9E + 05 |
| P-32 | 1.7E−02 | 1.7E + 01 | 1.7E + 04 |
| P-33 | 1.2E−01 | 1.2E + 02 | 1.2E + 05 |
| Pa-230 | 6.3E−04 | 6.3E−01 | 6.3E + 02 |
| Pa-231 | 8.3E−07 | 8.3E−04 | 8.3E−01 |
| Pa-233 | 9.3E−03 | 9.3E + 00 | 9.3E + 03 |
| Pa-234 | 9.3E−02 | 9.3E + 01 | 9.3E + 04 |
| Pb-203 | 8.3E−02 | 8.3E + 01 | 8.3E + 04 |
| Pb-205 | 1.2E−02 | 1.2E + 01 | 1.2E + 04 |
| Pb-209 | 1.1E + 01 | 1.1E + 04 | 1.1E + 07 |
| Pb-210 | 5.5E−05 | 5.5E−02 | 5.5E + 01 |
| Pb-211 | 1.2E−01 | 1.2E + 02 | 1.2E + 05 |
| Pb-212 | 6.0E−03 | 6.0E + 00 | 6.0E + 03 |
| Pb-214 | 1.2E−01 | 1.2E + 02 | 1.2E + 05 |
| Pd-103 | 2.1E−01 | 2.1E + 02 | 2.1E + 05 |
| Pd-107 | 8.2E−02 | 8.2E + 01 | 8.2E + 04 |
| Pd-109 | 9.4E−01 | 9.4E + 02 | 9.4E + 05 |
| Pm-143 | 7.6E−04 | 7.6E−01 | 7.6E + 02 |
| Pm-144 | 1.1E−04 | 1.1E−01 | 1.1E + 02 |
| Pm-145 | 5.2E−04 | 5.2E−01 | 5.2E + 02 |
| Pm-146 | 4.4E−05 | 4.4E−02 | 4.4E + 01 |
| Pm-147 | 2.6E−02 | 2.6E + 01 | 2.6E + 04 |
| Pm-148 | 1.7E−02 | 1.7E + 01 | 1.7E + 04 |
| Pm-148m | 7.6E−04 | 7.6E−01 | 7.6E + 02 |
| Pm-149 | 2.8E−01 | 2.8E + 02 | 2.8E + 05 |
| Pm-151 | 1.2E−01 | 1.2E + 02 | 1.2E + 05 |
| Po-210 | 9.3E−05 | 9.3E−02 | 9.3E + 01 |
| Pr-142 | 2.8E−01 | 2.8E + 02 | 2.8E + 05 |
| Pr-143 | 1.0E−01 | 1.0E + 02 | 1.0E + 05 |
| Pr-144 | 1.5E + 01 | 1.5E + 04 | 1.5E + 07 |
| Pt-191 | 6.4E−02 | 6.4E + 01 | 6.4E + 04 |
| Pt-193 | 2.1E−02 | 2.1E + 01 | 2.1E + 04 |
| Pt-193m | 4.8E−01 | 4.8E + 02 | 4.8E + 05 |
| Pt-195m | 1.4E−01 | 1.4E + 02 | 1.4E + 05 |
| Pt-197 | 1.1E + 00 | 1.1E + 03 | 1.1E + 06 |
| Pt-197m | 3.6E + 00 | 3.6E + 03 | 3.6E + 06 |
| Pu-236 | 7.0E−06 | 7.0E−03 | 7.0E + 00 |
| Pu-237 | 2.3E−02 | 2.3E + 01 | 2.3E + 04 |
| Pu-238 | 2.7E−06 | 2.7E−03 | 2.7E + 00 |
| Pu-239 | 2.5E−06 | 2.5E−03 | 2.5E + 00 |
| Pu-240 | 2.5E−06 | 2.5E−03 | 2.5E + 00 |
| Pu-241 | 1.3E−04 | 1.3E−01 | 1.3E + 02 |
| Pu-242 | 2.5E−06 | 2.5E−03 | 2.5E + 00 |
| Pu-243 | 3.8E + 00 | 3.8E + 03 | 3.8E + 06 |
| Pu-244 | 2.4E−06 | 2.4E−03 | 2.4E + 00 |
| Pu-245 | 2.1E−01 | 2.1E + 02 | 2.1E + 05 |
| Pu-246 | 4.8E−03 | 4.8E + 00 | 4.8E + 03 |
| Ra-223 | 1.3E−04 | 1.3E−01 | 1.3E + 02 |
| Ra-224 | 3.2E−04 | 3.2E−01 | 3.2E + 02 |
| Ra-225 | 1.3E−04 | 1.3E−01 | 1.3E + 02 |
| Ra-226 | 5.5E−06 | 5.5E−03 | 5.5E + 00 |
| Ra-228 | 1.3E−05 | 1.3E−02 | 1.3E + 01 |
| Rb-81 | 4.2E−01 | 4.2E + 02 | 4.2E + 05 |
| Rb-83 | 1.4E−03 | 1.4E + 00 | 1.4E + 03 |
| Rb-84 | 2.0E−03 | 2.0E + 00 | 2.0E + 03 |
| Rb-86 | 1.7E−02 | 1.7E + 01 | 1.7E + 04 |
| Rb-87 | 1.0E−02 | 1.0E + 01 | 1.0E + 04 |
| Rb-88 | 1.7E + 00 | 1.7E + 03 | 1.7E + 06 |
| Rb-89 | 6.4E−01 | 6.4E + 02 | 6.4E + 05 |
| Re−184 | 1.8E−03 | 1.8E + 00 | 1.8E + 03 |
| Re-184m | 3.6E−04 | 3.6E−01 | 3.6E + 02 |
| Re-186 | 1.9E−01 | 1.9E + 02 | 1.9E + 05 |
| Re-187 | 9.3E + 00 | 9.3E + 03 | 9.3E + 06 |
| Re-188 | 3.7E−01 | 3.7E + 02 | 3.7E + 05 |
| Rh-103m | 1.7E + 02 | 1.7E + 05 | 1.7E + 08 |
| Rh-105 | 3.4E−01 | 3.4E + 02 | 3.4E + 05 |
| Ru-97 | 8.3E−02 | 8.3E + 01 | 8.3E + 04 |
| Ru-103 | 3.1E−03 | 3.1E + 00 | 3.1E + 03 |
| Ru-105 | 2.9E−01 | 2.9E + 02 | 2.9E + 05 |
| Ru-106 | 5.9E−04 | 5.9E−01 | 5.9E + 02 |
| S-35 | 7.5E−02 | 7.5E + 01 | 7.5E + 04 |
| Sb-117 | 2.0E + 00 | 2.0E + 03 | 2.0E + 06 |
| Sb-122 | 3.9E−02 | 3.9E + 01 | 3.9E + 04 |
| Sb-124 | 6.0E−04 | 6.0E−01 | 6.0E + 02 |
| Sb-125 | 1.4E−04 | 1.4E−01 | 1.4E + 02 |
| Sb-126 | 1.8E−03 | 1.8E + 00 | 1.8E + 03 |
| Sb-126m | 7.6E−01 | 7.6E + 02 | 7.6E + 05 |
| Sb-127 | 2.0E−02 | 2.0E + 01 | 2.0E + 04 |
| Sb-129 | 1.8E−01 | 1.8E + 02 | 1.8E + 05 |
| Sc-44 | 1.4E−01 | 1.4E + 02 | 1.4E + 05 |
| Sc-46 | 4.0E−04 | 4.0E−01 | 4.0E + 02 |
| Sc-47 | 1.1E−01 | 1.1E + 02 | 1.1E + 05 |
| Sc-48 | 1.1E−02 | 1.1E + 01 | 1.1E + 04 |
| Sc-49 | 1.0E + 01 | 1.0E + 04 | 1.0E + 07 |
| Se-73 | 1.6E−01 | 1.6E + 02 | 1.6E + 05 |
| Se-75 | 1.1E−03 | 1.1E + 00 | 1.1E + 03 |
| Se-79 | 6.9E−03 | 6.9E + 00 | 6.9E + 03 |
| Si-31 | 4.7E + 00 | 4.7E + 03 | 4.7E + 06 |
| Si-32 | 7.2E−04 | 7.2E−01 | 7.2E + 02 |
| Sm-147 | 1.4E−05 | 1.4E−02 | 1.4E + 01 |
| Sm-151 | 3.5E−02 | 3.5E + 01 | 3.5E + 04 |
| Sm-153 | 2.4E−01 | 2.4E + 02 | 2.4E + 05 |
| Sn-113 | 1.9E−03 | 1.9E + 00 | 1.9E + 03 |
| Sn-117m | 2.3E−02 | 2.3E + 01 | 2.3E + 04 |
| Sn-119m | 2.8E−02 | 2.8E + 01 | 2.8E + 04 |
| Sn-123 | 1.8E−02 | 1.8E + 01 | 1.8E + 04 |
| Sn-125 | 7.2E−03 | 7.2E + 00 | 7.2E + 03 |
| Sn-126 | 4.7E−06 | 4.7E−03 | 4.7E + 00 |
| Sr-82 | 1.9E−03 | 1.9E + 00 | 1.9E + 03 |
| Sr-85 | 1.9E−03 | 1.9E + 00 | 1.9E + 03 |
| Sr-85m | 1.5E + 00 | 1.5E + 03 | 1.5E + 06 |
| Sr-87m | 1.2E + 00 | 1.2E + 03 | 1.2E + 06 |
| Sr-89 | 2.1E−02 | 2.1E + 01 | 2.1E + 04 |
| Sr-90 | 5.2E−04 | 5.2E−01 | 5.2E + 02 |
| Sr-91 | 1.2E−01 | 1.2E + 02 | 1.2E + 05 |
| Sr-92 | 2.5E−01 | 2.5E + 02 | 2.5E + 05 |
| Ta-182 | 4.4E−04 | 4.4E−01 | 4.4E + 02 |
| Tb-157 | 2.2E−03 | 2.2E + 00 | 2.2E + 03 |
| Tb-160 | 8.4E−04 | 8.4E−01 | 8.4E + 02 |
| Tc-95 | 9.0E−02 | 9.0E + 01 | 9.0E + 04 |
| Tc-95m | 1.4E−03 | 1.4E + 00 | 1.4E + 03 |
| Tc-96 | 5.6E−03 | 5.6E + 00 | 5.6E + 03 |
| Tc-96m | 7.0E−01 | 7.0E + 02 | 7.0E + 05 |
| Tc-97 | 1.5E−03 | 1.5E + 00 | 1.5E + 03 |
| Tc-97m | 7.2E−02 | 7.2E + 01 | 7.2E + 04 |
| Tc-98 | 6.4E−06 | 6.4E−03 | 6.4E + 00 |
| Tc-99 | 9.0E−03 | 9.0E + 00 | 9.0E + 03 |
| Tc-99m | 1.4E + 00 | 1.4E + 03 | 1.4E + 06 |
| Tc-101 | 3.8E + 00 | 3.8E + 03 | 3.8E + 06 |
| Te-121 | 6.0E−03 | 6.0E + 00 | 6.0E + 03 |
| Te-121m | 5.3E−04 | 5.3E−01 | 5.3E + 02 |
| Te-123 | 1.2E−03 | 1.2E + 00 | 1.2E + 03 |
| Te-123m | 2.7E−03 | 2.7E + 00 | 2.7E + 03 |
| Te-125m | 1.5E−02 | 1.5E + 01 | 1.5E + 04 |
| Te-127 | 2.9E + 00 | 2.9E + 03 | 2.9E + 06 |
| Te-127m | 7.3E−03 | 7.3E + 00 | 7.3E + 03 |
| Te-129 | 6.5E + 00 | 6.5E + 03 | 6.5E + 06 |
| Te-129m | 6.1E−03 | 6.1E + 00 | 6.1E + 03 |
| Te-131 | 9.4E−01 | 9.4E + 02 | 9.4E + 05 |
| Te-131m | 1.8E−02 | 1.8E + 01 | 1.8E + 04 |
| Te-132 | 6.2E−03 | 6.2E + 00 | 6.2E + 03 |
| Te-133 | 1.2E + 00 | 1.2E + 03 | 1.2E + 06 |
| Te-133m | 2.9E−01 | 2.9E + 02 | 2.9E + 05 |
| Te-134 | 4.4E−01 | 4.4E + 02 | 4.4E + 05 |
| Th-226 | 3.0E−02 | 3.0E + 01 | 3.0E + 04 |
| Th-227 | 6.4E−05 | 6.4E−02 | 6.4E + 01 |
| Th-228 | 2.9E−06 | 2.9E−03 | 2.9E + 00 |
| Th-229 | 4.9E−07 | 4.9E−04 | 4.9E−01 |
| Th-230 | 3.2E−06 | 3.2E−03 | 3.2E + 00 |
| Th-231 | 8.4E−01 | 8.4E + 02 | 8.4E + 05 |
| Th-232 | 6.0E−07 | 6.0E−04 | 6.0E−01 |
| Th-234 | 2.0E−02 | 2.0E + 01 | 2.0E + 04 |
| Ti-44 | 5.2E−06 | 5.2E−03 | 5.2E + 00 |
| Ti-45 | 4.0E−01 | 4.0E + 02 | 4.0E + 05 |
| Tl-200 | 4.4E−02 | 4.4E + 01 | 4.4E + 04 |
| Tl-201 | 1.8E−01 | 1.8E + 02 | 1.8E + 05 |
| Tl-202 | 1.0E−02 | 1.0E + 01 | 1.0E + 04 |
| Tl-204 | 2.5E−02 | 2.5E + 01 | 2.5E + 04 |
| Tm-170 | 2.4E−02 | 2.4E + 01 | 2.4E + 04 |
| Tm-171 | 5.9E−02 | 5.9E + 01 | 5.9E + 04 |
| U-230 | 5.0E−05 | 5.0E−02 | 5.0E + 01 |
| U-231 | 1.4E−01 | 1.4E + 02 | 1.4E + 05 |
| U-232 | 1.3E−06 | 1.3E−03 | 1.3E + 00 |
| U-233 | 7.6E−06 | 7.6E−03 | 7.6E + 00 |
| U-234 | 7.6E−06 | 7.6E−03 | 7.6E + 00 |
| U-235 | 7.0E−06 | 7.0E−03 | 7.0E + 00 |
| U-236 | 8.4E−06 | 8.4E−03 | 8.4E + 00 |
| U-237 | 4.7E−02 | 4.7E + 01 | 4.7E + 04 |
| U-238 | 8.6E−06 | 8.6E−03 | 8.6E + 00 |
| U-239 | 8.3E + 00 | 8.3E + 03 | 8.3E + 06 |
| U-240 | 1.8E−01 | 1.8E + 02 | 1.8E + 05 |
| V-48 | 1.4E−03 | 1.4E + 00 | 1.4E + 03 |
| V-49 | 1.3E + 00 | 1.3E + 03 | 1.3E + 06 |
| W-181 | 1.1E−02 | 1.1E + 01 | 1.1E + 04 |
| W-185 | 1.6E−01 | 1.6E + 02 | 1.6E + 05 |
| W-187 | 1.1E−01 | 1.1E + 02 | 1.1E + 05 |
| W-188 | 1.0E−02 | 1.0E + 01 | 1.0E + 04 |
| Xe-122 | 7.6E−02 | 7.6E + 01 | 7.6E + 04 |
| Xe-123 | 1.6E + 00 | 1.6E + 03 | 1.6E + 06 |
| Xe-125 | 6.0E−01 | ||
| Xe-127 | 7.0E + 00 | ||
| Xe-129m | 7.6E + 01 | ||
| Xe-131m | 2.2E + 02 | ||
| Xe-133 | 5.2E + 01 | ||
| Xe-133m | 6.0E + 01 | ||
| Xe-135 | 7.6E + 00 | ||
| Xe-135m | 4.2E + 00 | ||
| Xe-138 | 9.9E−01 | ||
| Y-86 | 2.8E−02 | 2.8E + 01 | 2.8E + 04 |
| Y-87 | 2.3E−02 | 2.3E + 01 | 2.3E + 04 |
| Y-88 | 2.5E−04 | 2.5E−01 | 2.5E + 02 |
| Y-90 | 1.1E−01 | 1.1E + 02 | 1.1E + 05 |
| Y-90m | 4.3E−01 | 4.3E + 02 | 4.3E + 05 |
| Y-91 | 1.8E−02 | 1.8E + 01 | 1.8E + 04 |
| Y-91m | 1.6E + 00 | 1.6E + 03 | 1.6E + 06 |
| Y-92 | 7.0E−01 | 7.0E + 02 | 7.0E + 05 |
| Y-93 | 3.8E−01 | 3.8E + 02 | 3.8E + 05 |
| Yb-169 | 5.5E−03 | 5.5E + 00 | 5.5E + 03 |
| Yb-175 | 2.1E−01 | 2.1E + 02 | 2.1E + 05 |
| Zn-62 | 8.6E−02 | 8.6E + 01 | 8.6E + 04 |
| Zn-65 | 4.4E−04 | 4.4E−01 | 4.4E + 02 |
| Zn-69 | 2.7E + 01 | 2.7E + 04 | 2.7E + 07 |
| Zn-69m | 2.0E−01 | 2.0E + 02 | 2.0E + 05 |
| Zr-86 | 2.4E−02 | 2.4E + 01 | 2.4E + 04 |
| Zr-88 | 2.7E−04 | 2.7E−01 | 2.7E + 02 |
| Zr-89 | 1.6E−02 | 1.6E + 01 | 1.6E + 04 |
| Zr-93 | 2.8E−03 | 2.8E + 00 | 2.8E + 03 |
| Zr-95 | 6.4E−04 | 6.4E−01 | 6.4E + 02 |
| Zr-97 | 4.6E−02 | 4.6E + 01 | 4.6E + 04 |
* Radionuclides boiling at 100 °C or less, or exposed to a temperature of 100 °C, must be considered a gas. Capsules containing radionuclides in liquid or powder form can be considered to be solids.
** Mo-99 contained in a generator to produce Technetium-99 can be assumed to be a solid.
(a) Table 2 may be used for determining if facilities are in compliance with the standard.
1. The concentration table as applied to emission estimates can only be used if all releases are from point sources and concentrations have been measured at the stack or vent using EPA-approved methods, and the distance between each stack or vent and the nearest resident is greater than 3 times the diameter of the stack or vent. Procedures provided in Ref. (1) shall be used to determine compliance or exemption from reporting by use of Table 2.
2. The concentration table may be used to determine compliance with the standard based on environmental measurements provided these measurements are made in conformance with the requirements of § 61.107(b)(5).
4. NCRP Screening ModelThe procedures described in Reference (4) may be used to determine doses to members of the general public from emissions of radionuclides to the atmosphere. Both the total dose from all radionuclides emitted, and the dose caused by radioactive iodine must be considered in accordance with the procedures in Ref. (1).
5. The COMPLY Computer CodeThe COMPLY computer code may be used to determine compliance with subpart I. The compliance model in the COMPLY computer code may be used to determine the dose to members of the general public from emissions of radionuclides to the atmosphere. The EPA may add radionuclides to all or any part of COMPLY to cover radionuclides that may be used by the regulated community.
Table 2 - Concentration Levels for Environmental Compliance
| Radionuclide | Concentration (Ci/m 3) | Radionuclide | Concentration (Ci/m 3) |
|---|---|---|---|
| Ac-225 | 9.1E−14 | Bi-207 | 1.0E−14 |
| Ac-227 | 1.6E−16 | Bi-210 | 2.9E−13 |
| Ac-228 | 3.7E−12 | Bi-212 | 5.6E−11 |
| Ag-106 | 1.9E−09 | Bi-213 | 7.1E−11 |
| Ag-106m | 1.2E−12 | Bi-214 | 1.4E−10 |
| Ag-108m | 7.1E−15 | Bk-249 | 5.6E−13 |
| Ag-110m | 9.1E−14 | Bk-250 | 9.1E−11 |
| Ag-111 | 2.5E−12 | Br-77 | 4.2E−11 |
| Al-26 | 4.8E−15 | Br-80 | 1.4E−08 |
| Am-241 | 1.9E−15 | Br-80m | 1.8E−09 |
| Am-242 | 1.5E−11 | Br-82 | 1.2E−11 |
| Am-242m | 2.0E−15 | Br-83 | 1.2E−08 |
| Am-243 | 1.8E−15 | Br-84 | 6.7E−10 |
| Am-244 | 4.0E−11 | C-11 | 1.5E−09 |
| Am-245 | 8.3E−09 | C-14 | 1.0E−11 |
| Am-246 | 1.2E−09 | Ca-41 | 4.2E−13 |
| Ar-37 | 1.6E−03 | Ca-45 | 1.3E−12 |
| Ar-41 | 1.7E−09 | Ca-47 | 2.4E−12 |
| As-72 | 2.4E−11 | Cd-109 | 5.9E−13 |
| As-73 | 1.1E−11 | Cd-113 | 9.1E−15 |
| As-74 | 2.2E−12 | Cd-113m | 1.7E−14 |
| As-76 | 5.0E−11 | Cd-115 | 1.6E−11 |
| As-77 | 1.6E−10 | Cd-115m | 8.3E−13 |
| At-211 | 1.1E−11 | Cd-117 | 6.7E−11 |
| Au-193 | 3.8E−10 | Cd-117m | 1.6E−10 |
| Au-194 | 3.2E−11 | Ce-139 | 2.6E−12 |
| Au-195 | 3.1E−12 | Ce-141 | 6.3E−12 |
| Au-198 | 2.1E−11 | Ce-143 | 3.0E−11 |
| Au-199 | 4.8E−11 | Ce-144 | 6.2E−13 |
| Ba-131 | 7.1E−12 | Cf-248 | 1.8E−14 |
| Ba-133 | 5.9E−14 | Cf-249 | 1.4E−15 |
| Ba-133m | 5.9E−11 | Cf-250 | 3.2E−15 |
| Ba-135m | 1.8E−10 | Cf-251 | 1.4E−15 |
| Ba-139 | 5.6E−09 | Cf-252 | 5.6E−15 |
| Ba-140 | 1.3E−12 | Cf-253 | 3.1E−13 |
| Ba-141 | 1.4E−09 | Cf-254 | 3.0E−15 |
| Ba-142 | 1.3E−09 | Cl-36 | 2.7E−15 |
| Be-7 | 2.3E−11 | Cl-38 | 7.7E−10 |
| Be-10 | 1.6E−12 | Cm-242 | 5.3E−14 |
| Bi-206 | 2.3E−12 | Cm-243 | 2.6E−15 |
| Cm-244 | 3.3E−15 | Eu-156 | 1.9E−12 |
| Cm-245 | 1.8E−15 | F-18 | 6.7E−10 |
| Cm-246 | 1.9E−15 | Fe-52 | 5.6E−11 |
| Cm-247 | 1.9E−15 | Fe-55 | 9.1E−12 |
| Cm-248 | 5.0E−16 | Fe-59 | 6.7E−13 |
| Cm-249 | 3.7E−09 | Fm-254 | 2.0E−11 |
| Cm-250 | 9.1E−17 | Fm-255 | 4.3E−12 |
| Co-56 | 1.8E−13 | Fr-223 | 3.3E−11 |
| Co-57 | 1.3E−12 | Ga-66 | 6.2E−11 |
| Co-58 | 6.7E−13 | Ga-67 | 7.1E−11 |
| Co-58m | 1.2E−10 | Ga-68 | 9.1E−10 |
| Co-60 | 1.7E−14 | Ga-72 | 3.8E−11 |
| Co-60m | .4.3E−09 | Gd-152 | 5.0E−15 |
| Co-61 | 4.5E−09 | Gd-153 | 2.1E−12 |
| Cr-49 | 1.1E−09 | Gd-159 | 2.9E−10 |
| Cr-51 | 3.1E−11 | Ge-68 | 2.0E−13 |
| Cs-129 | 1.4E−10 | Ge-71 | 2.4E−10 |
| Cs-131 | 3.3E−11 | Ge-77 | 1.0E−10 |
| Cs-132 | 4.8E−12 | H-3 | 1.5E−09 |
| Cs-134 | 2.7E−14 | Hf-181 | 1.9E−12 |
| Cs-134m | 1.7E−10 | Hg-193m | 1.0E−10 |
| Cs-135 | 4.0E−13 | Hg-197 | 8.3E−11 |
| Cs-136 | 5.3E−13 | Hg-197m | 1.1E−10 |
| Cs-137 | 1.9E−14 | Hg-203 | 1.0E−12 |
| Cs-138 | 5.3E−10 | Ho-166 | 7.1E−11 |
| Cu-61 | 4.8E−10 | Ho-166m | 7.1E−15 |
| Cu-64 | 5.3E−10 | I-123 | 4.3E−10 |
| Cu-67 | 5.0E−11 | I-124 | 6.2E−13 |
| Dy-157 | 5.0E−10 | I-125 | 1.2E−13 |
| Dy-165 | 6.7E−09 | I-126 | 1.1E−13 |
| Dy-166 | 1.1E−11 | I-128 | 1.1E−08 |
| Er-169 | 2.9E−11 | I-129 | 9.1E−15 |
| Er-171 | 4.0E−10 | I-130 | 4.5E−11 |
| Es-253 | 2.4E−13 | I-131 | 2.1E−13 |
| Es-254 | 2.0E−14 | I-132 | 2.3E−10 |
| Es-254m | 1.8E−12 | I-133 | 2.0E−11 |
| Eu-152 | 2.0E−14 | I-134 | 3.8E−10 |
| Eu-152m | 3.6E−10 | I-135 | 1.2E−10 |
| Eu-154 | 2.3E−14 | In-111 | 3.6E−11 |
| Eu-155 | 5.9E−13 | In-113m | 2.5E−09 |
| In-114m | 9.1E−13 | Nb-95 | 2.2E−12 |
| In-115 | 7.1E−14 | Nb-95m | 1.4E−11 |
| In-115m | 1.6E−09 | Nb-96 | 2.4E−11 |
| In-116m | 4.2E−10 | Nb-97 | 1.2E−09 |
| In-117 | 1.6E−09 | Nd-147 | 7.7E−12 |
| In-117m | 9.1E−11 | Nd-149 | 7.1E−10 |
| Ir-190 | 2.6E−12 | Ni-56 | 1.7E−12 |
| Ir-192 | 9.1E−13 | Ni-57 | 1.8E−11 |
| Ir-194 | 1.1E−10 | Ni-59 | 1.5E−11 |
| Ir-194m | 1.7E−13 | Ni-63 | 1.4E−11 |
| K-40 | 2.7E−14 | Ni-65 | 8.3E−10 |
| K-42 | 2.6E−10 | Np-235 | 2.5E−11 |
| K-43 | 6.2E−11 | Np-237 | 1.2E−15 |
| K-44 | 5.9E−10 | Np-238 | 1.4E−11 |
| Kr-79 | 8.3E−09 | Np-239 | 3.8E−11 |
| Kr-81 | 2.1E−07 | Np-240 | 7.7E−10 |
| Kr-83m | 2.3E−05 | Np-240m | 5.6E−09 |
| Kr-85 | 1.0E−06 | Os-185 | 1.0E−12 |
| Kr-85m | 1.3E−08 | Os-191m | 2.9E−10 |
| Kr-87 | 2.4E−09 | Os-191 | 1.1E−11 |
| Kr-88 | 5.0E−10 | Os-193 | 9.1E−11 |
| La-140 | 1.2E−11 | P-32 | 3.3E−13 |
| La-141 | 7.7E−10 | P-33 | 2.4E−12 |
| La-142 | 2.7E−10 | Pa-230 | 3.2E−13 |
| Lu-177 | 2.4E−11 | Pa-231 | 5.9E−16 |
| Lu-177m | 3.6E−13 | Pa-233 | 4.8E−12 |
| Mg-28 | 1.5E−11 | Pa-234 | 1.1E−10 |
| Mn-52 | 2.8E−12 | Pb-203 | 6.2E−11 |
| Mn-52m | 6.2E−10 | Pb-205 | 5.6E−12 |
| Mn-53 | 1.5E−11 | Pb-209 | 1.3E−08 |
| Mn-54 | 2.8E−13 | Pb-2I0 | 2.8E−15 |
| Mn-56 | 2.9E−10 | Pb-211 | 1.4E−10 |
| Mo-93 | 1.1E−12 | Pb-212 | 6.3E−12 |
| Mo-99 | 1.4E−11 | Pb-214 | 1.2E−10 |
| Mo-101 | 1.0E−09 | Pd-103 | 3.8E−11 |
| Na-22 | 2.6E−14 | Pd-107 | 3.1E−11 |
| Na-24 | 2.6E−11 | Pd-109 | 4.8E−10 |
| Nb-90 | 2.6E−11 | Pm-143 | 9.1E−13 |
| Nb-93m | 1.0E−11 | Pm-144 | 1.3E−13 |
| Nb-94 | 7.1E−15 | Pm-145 | 6.2E−13 |
| Pm-146 | 5.3E−14 | Re-184m | 3.7E−13 |
| Pm-147 | 1.1E−11 | Re-186 | 1.8E−11 |
| Pm-148 | 5.0E−12 | Re-187 | 2.6E−10 |
| Pm-148m | 6.7E−13 | Re-188 | 1.7E−10 |
| Pm-149 | 4.2E−11 | Rh-103m | 2.1E−07 |
| Pm-151 | 7.1E−11 | Rh-105 | 1.3E−10 |
| Po-210 | 7.1E−15 | Ru-97 | 6.7E−11 |
| Pr-142 | 1.1E−10 | Ru-103 | 2.6E−12 |
| Pr-143 | 7.1E−12 | Ru-105 | 2.8E−10 |
| Pr-144 | 1.8E−08 | Ru-106 | 3.4E−13 |
| Pt-191 | 4.3E−11 | S-35 | 1.3E−12 |
| Pt-193 | 1.8E−11 | Sb-117 | 2.4E−09 |
| Pt-193m | 4.8E−11 | Sb-122 | 1.4E−11 |
| Pt-195m | 3.2E−11 | Sb-124 | 5.3E−13 |
| Pt-197 | 4.0E−10 | Sb-125 | 1.6E−13 |
| Pt-197m | 2.6E−09 | Sb-126 | 1.4E−12 |
| Pu-236 | 5.9E−15 | Sb-126m | 9.1E−10 |
| Pu-237 | 1.9E−11 | Sb-127 | 7.1E−12 |
| Pu-238 | 2.1E−15 | Sb-129 | 7.7E−11 |
| Pu-239 | 2.0E−15 | Sc-44 | 1.7E−10 |
| Pu-240 | 2.0E−15 | Sc-46 | 4.2E−13 |
| Pu-241 | 1.0E−13 | Sc-47 | 3.8E−11 |
| Pu-242 | 2.0E−15 | Sc-48 | 9.1E−12 |
| Pu-243 | 4.2E−09 | Sc-49 | 1.2E−08 |
| Pu-244 | 2.0E−15 | Se-73 | 1.7E−10 |
| Pu-245 | 2.1E−10 | Se-75 | 1.7E−13 |
| Pu-246 | 2.2E−12 | Se-79 | 1.1E−13 |
| Ra-223 | 4.2E−14 | Si-31 | 5.6E−09 |
| Ra-224 | 1.5E−13 | Si-32 | 3.4E−14 |
| Ra-225 | 5.0E−14 | Sm-147 | 1.4E−14 |
| Ra-226 | 3.3E−15 | Sm-151 | 2.1E−11 |
| Ra-228 | 5.9E−15 | Sm-153 | 5.9E−11 |
| Rb-81 | 5.0E−10 | Sn-113 | 1.4E−12 |
| Rb-83 | 3.4E−13 | Sn-117m | 5.6E−12 |
| Rb-84 | 3.6E−13 | Sn-119m | 5.3E−12 |
| Rb-86 | 5.6E−13 | Sn-123 | 1.1E−12 |
| Rb-87 | 1.6E−13 | Sn-125 | 1.7E−12 |
| Rb-88 | 2.1E−09 | Sn-126 | 5.3E−15 |
| Rb-89 | 7.1E−10 | Sr-82 | 6.2E−13 |
| Re-184 | 1.5E−12 | Sr-85 | 1.8E−12 |
| Sr-85m | 1.6E−09 | Th-232 | 6.2E−16 |
| Sr-87m | 1.4E−09 | Th-234 | 2.2E−12 |
| Sr-89 | 1.8E−12 | Ti-44 | 6.2E−15 |
| Sr-90 | 1.9E−14 | Ti-45 | 4.8E−10 |
| Sr-91 | 9.1E−11 | Tl-200 | 4.5E−11 |
| Sr-92 | 2.9E−10 | Tl-201 | 1.0E−10 |
| Ta-182 | 4.5E−13 | Tl-202 | 5.0E−12 |
| Tb-157 | 2.5E−12 | Tl-204 | 1.2E−12 |
| Tb-160 | 7.7E−13 | Tm-170 | 3.3E−12 |
| Tc-95 | 1.0E−10 | Tm-171 | 2.6E−11 |
| Tc-95m | 1.4E−12 | U-230 | 1.5E−14 |
| Tc-96 | 5.6E−12 | U-231 | 4.2E−11 |
| Tc-96m | 6.7E−10 | U-232 | 1.3E−15 |
| Tc-97 | .7.1E−13 | U-233 | 7.1E−15 |
| Tc-97m | 7.1E−12 | U-234 | 7.7E−15 |
| Tc-98 | 6.7E−15 | U-235 | 7.1E−15 |
| Tc-99 | 1.4E−13 | U-236 | 7.7E−15 |
| Tc-99m | 1.7E−09 | U-237 | 1.0E−11 |
| Tc-101 | 4.5E−09 | U-238 | 8.3E−15 |
| Te-121 | 1.0E−12 | U-239 | 4.3E−09 |
| Te-121m | 1.2E−13 | U-240 | 1.3E−10 |
| Te-123 | 1.4E−13 | V-48 | 1.0E−12 |
| Te-123m | 2.0E−13 | V-49 | 1.6E−10 |
| Te-125m | 3.6E−13 | W-181 | 6.7E−12 |
| Te-127 | 1.0E−09 | W-185 | 2.6E−12 |
| Te-127m | 1.5E−13 | W-187 | 7.7E−11 |
| Te-129 | 7.7E−09 | W-188 | 5.3E−13 |
| Te-129m | 1.4E−13 | Xe-122 | 9.1E−11 |
| Te-131 | 9.1E−11 | Xe-123 | 1.6E−09 |
| Te-131m | 1.0E−12 | Xe-125 | 1.1E−11 |
| Te-132 | 7.1E−13 | Xe-127 | 8.3E−09 |
| Te-133 | 9.1E−10 | Xe-129m | 9.1E−08 |
| Te-133m | 2.2E−10 | Xe-131m | 2.6E−07 |
| Te-134 | 5.3E−10 | Xe-133 | 6.2E−08 |
| Th-226 | 3.4E−11 | Xe-133m | 7.1E−08 |
| Th-227 | 3.8E−14 | Xe-135 | 9.1E−09 |
| Th-228 | 3.1E−15 | Xe-135m | 5.0E−09 |
| Th-229 | 5.3E−16 | Xe-138 | 1.2E−09 |
| Th-230 | 3.4E−15 | Y-86 | 3.0E−11 |
| Th-231 | 2.9E−10 | Y-87 | 1.7E−11 |
| Y-88 | 2.7E−13 | Zn-65 | 9.1E−14 |
| Y-90 | 1.3E−11 | Zn-69 | 3.2E−08 |
| Y-90m | 1.9E−10 | Zn-69m | 1.7E−10 |
| Y-91 | 2.1E−12 | Zr-86 | 2.4E−11 |
| Y-91m | 1.3E−09 | Zr-88 | 3.1E−13 |
| Y-92 | 8.3E−10 | Zr-89 | 1.3E−11 |
| Y-93 | 2.9E−10 | Zr-93 | 2.6E−12 |
| Yb-169 | 3.7E−12 | Zr-95 | 6.7E−13 |
| Yb-175 | 4.3E−11 | Zr-97 | 3.8E−11 |
| Zn-62 | 9.1E−11 |
(1) Environmental Protection Agency, “A Guide for Determining Compliance with the Clean Air Act Standards for Radionuclides Emissions from NRC-Licensed and Non-DOE Federal Facilities”, EPA 520/1-89-002, October 1989.
(2) Environmental Protection Agency, “User's Guide for the COMPLY Code”, EPA 520/1-89-003, October 1989.
(3) Environmental Protection Agency, “Background Information Document: Procedures Approved for Demonstrating Compliance with 40 CFR Part 61, Subpart I”, EPA 520/1-89-001, January 1989.
(4) National Council on Radiation Protection and Measurement, “Screening Techniques for Determining Compliance with Environmental Standards” NCRP Commentary No. 3, Revision of January 1989 with addendum of October, 1989.
[54 FR 51711, Dec. 15, 1989]