Title 40
PART 435 APPENDIX
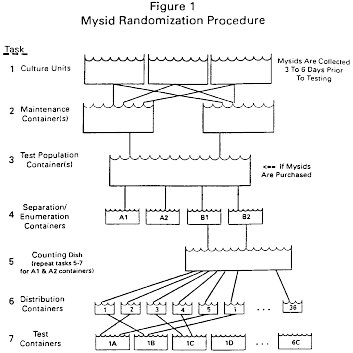
| Selection/assignment round (2 mysids each) | Place mysid in the numbered distribution containers in the random order shown |
|---|---|
| 1 | 8, 21, 6, 28, 33, 32, 1, 3, 10, 9, 4, 14, 23, 2, 34, 22, 36, 27, 5, 30, 35, 24, 12, 25, 11, 17, 19, 26, 31, 7, 20, 15, 18, 13, 16, 29. |
| 2 | 35, 18, 5, 12, 32, 34, 22, 3, 9, 16, 26, 13, 20, 28, 6, 21, 24, 30, 8, 31, 7, 23, 2, 15, 25, 17, 1, 11, 27, 4, 19, 36, 10, 33, 14, 29. |
| 3 | 7, 19, 14, 11, 34, 21, 25, 27, 17, 18, 6, 16, 29, 2, 32, 10, 4, 20, 3, 9, 1, 5, 28, 24, 31, 15, 22, 13, 33, 26, 36, 12, 8, 30, 35, 23. |
| 4 | 30, 2, 18, 5, 8, 27, 10, 25, 4, 20, 26, 15, 31, 36, 35, 23, 11, 29, 16, 17, 28, 1, 33, 14, 9, 34, 7, 3, 12, 22, 21, 6, 19, 24, 32, 13. |
| 5 | 34, 28, 16, 17, 10, 12, 1, 36, 20, 18, 15, 22, 2, 4, 19, 23, 27, 29, 25, 21, 30, 3, 9, 33, 32, 6, 14, 11, 35, 24, 26, 7, 31, 5, 13, 8. |
(8) Transfer mysids from the 36 distribution containers to 18 labeled test containers in random order. A label is assigned to each of the three replicates (A, B, C) of the six test concentrations. Count and record the 96 hour response in an impartial order.
(9) Repeat tasks 5-7 for each toxicity test. A new random schedule should be followed in Tasks 6 and 7 for each test.
Note:If a partial toxicity test is conducted, the procedures described above are appropriate and should be used to prepare the single test concentration and control, along with the reference toxicant test.
V-B. Data Analysis and Interpretation(1) Complete survival data in all test containers at each observation time shall be presented in tabular form. If greater than 10 percent mortality occurs in the controls, all data shall be discarded and the experiment repeated. Unacceptably high control mortality indicates the presence of important stresses on the organisms other than the material being tested, such as injury or disease, stressful physical or chemical conditions in the containers, or improper handling, acclimation, or feeding. If 10 percent mortality or less occurs in the controls, the data may be evaluated and reported.
(2) A definitive, full bioassay conducted according to the EPA protocol is used to estimate the concentration that is lethal to 50 percent of the test organisms that do not die naturally. This toxicity measure is known as the median lethal concentration, or LC-50. The LC-50 is adjusted for natural mortality or natural responsiveness. The maximum likelihood estimation procedure with the adjustments for natural responsiveness as given by D.J. Finney, in Probit Analysis 3rd edition, 1971, Cambridge University Press, chapter 7, can be used to obtain the probit model estimate of the LC-50 and the 95 percent fiducial (confidence) limits for the LC-50. These estimates are obtained using the logarithmic transform of the concentration. The heterogeneity factor (Finney 1971, pages 70-72) is not used. For a test material to pass the toxicity test, according to the requirements stated in the offshore oil and gas extraction industry BAT effluent limitations and NSPS, the LC-50, adjusted for natural responsiveness, must be greater than 3 percent suspended particulate phase (SPP) concentration by volume unadjusted for the 1 to 9 dilution. Other toxicity test models may be used to obtain toxicity estimates provided the modeled mathematical expression for the lethality rate must increase continuously with concentration. The lethality rate is modeled to increase with concentration to reflect an assumed increase in toxicity with concentration even though the observed lethality may not increase uniformly because of the unpredictable animal response fluctuations.
(3) The range finding test is used to establish a reasonable set of test concentrations in order to run the definitive test. However, if the lethality rate changes rapidly over a narrow range of concentrations, the range finding assay may be too coarse to establish an adequate set of test concentrations for a definitive test.
(4) The EPA Environmental Research Laboratory in Gulf Breeze, Florida prepared a Research and Development Report entitled Acute Toxicity of Eight Drilling Fluids to Mysid Shrimp (Mysidopsis bahia), May 1984 EPA-600/3-84-067. The Gulf Breeze data for drilling fluid number 1 are displayed in Table 1 for purposes of an example of the probit analysis described above. The SAS Probit Procedure (SAS Institute, Statistical Analysis System, Cary, North Carolina, 1982) was used to analyze these data. The 96-hour LC50 adjusted for the estimated spontaneous mortality rate is 3.3 percent SPP with 95 percent limits of 3.0 and 3.5 percent SPP with the 1 to 9 dilution. The estimated spontaneous mortality rate based on all of the data is 9.6 percent.
Table 1 - Listing of Acute Toxicity Test Data (August 1983 to September 1983) with Eight Generic Drilling Fluids and Mysid Shrimp
[fluid N2 = 1]
| Percent concentration | Number exposed | Number dead (96 hours) | Number alive (96 hours) |
|---|---|---|---|
| 0 | 60 | 3 | 57 |
| 1 | 60 | 11 | 49 |
| 2 | 60 | 11 | 49 |
| 3 | 60 | 25 | 35 |
| 4 | 60 | 48 | 12 |
| 5 | 60 | 60 | 0 |
(1) A partial test conducted according to EPA protocol can be used economically to demonstrate that a test material passes the toxicity test. The partial test cannot be used to estimate the LC-50 adjusted for natural response.
(2) To conduct a partial test follow the test protocol for preparation of the test material and organisms. Prepare the control (zero concentration), one test concentration (3 percent suspended particulate phase) and the reference toxicant according to the methods of the full test. A range finding test is not used for the partial test.
(3) Sixty test organisms are used for each test concentration. Find the number of test organisms killed in the control (zero percent SPP) concentration in the column labeled X0 of Table 2. If the number of organisms in the control (zero percent SPP) exceeds the table values, then the test is unacceptable and must be repeated. If the number of organisms killed in the 3 percent test concentration is less than or equal to corresponding number in the column labeled X1 then the test material passes the partial toxicity test. Otherwise the test material fails the toxicity test.
(4) Data shall be reported as percent suspended particulate phase.
Table 2
| X0 | X1 |
|---|---|
| 0 | 22 |
| 1 | 22 |
| 2 | 23 |
| 3 | 23 |
| 4 | 24 |
| 5 | 24 |
| 6 | 25 |
1. Borthwick, Patrick W. 1978. Methods for acute static toxicity tests with mysid shrimp (Mysidopsis bahia). Bioassay Procedures for the Ocean Disposal Permit Program, [EPA-600/9-78-010:] March.
2. Nimmo, D.R., T.L. Hamaker, and C.A. Somers. 1978. Culturing the mysid (Mysidopsis bahia) in flowing seawater or a static system. Bioassay Procedures for the Ocean Disposal Permit Program, [EPA-600/9-78-010]: March.
3. American Public Health Association et al. 1980. Standard Methods for the Examination of Water and Wastewater. Washington, DC, 15th Edition: 90-99.
4. U.S. Environmental Protection Agency, September 1991. Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms. EPA/600/4-90/027. Washington, DC, 4th Edition.
5. Finney, D.J. Probit Analysis. Cambridge University Press; 1971.
6. U.S. Environmental Protection Agency, May 1984. Acute Toxicity of Eight Drilling Fluids to Mysid Shrimp (Mysidopsis bahia). EPA-600/3-84-067.
[58 FR 12504, Mar. 4, 1993, as amended at 77 FR 29837, May 18, 2012]Appendix 3 to Subpart A of Part 435 - Procedure for Mixing Base Fluids With Sediments (EPA Method 1646)
40:32.0.1.1.11.1.4.7.9 :
Appendix 3 to Subpart A of Part 435 - Procedure for Mixing Base Fluids With Sediments (EPA Method 1646)This procedure describes a method for amending uncontaminated and nontoxic (control) sediments with the base fluids that are used to formulate synthetic-based drilling fluids and other non-aqueous drilling fluids. Initially, control sediments shall be press-sieved through a 2000 micron mesh sieve to remove large debris. Then press-sieve the sediment through a 500 micron sieve to remove indigenous organisms that may prey on the test species or otherwise confound test results. Homogenize control sediment to limit the effects of settling that may have occurred during storage. Sediments should be homogenized before density determinations and addition of base fluid to control sediment. Because base fluids are strongly hydrophobic and do not readily mix with sediment, care must be taken to ensure base fluids are thoroughly homogenized within the sediment. All concentrations are weight-to-weight (mg of base fluid to kg of dry control sediment). Sediment and base fluid mixing shall be accomplished by using the following method.
1. Determine the wet to dry ratio for the control sediment by weighing approximately 10 g subsamples of the screened and homogenized wet sediment into tared aluminum weigh pans. Dry sediment at 105 °C for 18-24 h. Remove sediment and cool in a desiccator until a constant weight is achieved. Re-weigh the samples to determine the dry weight. Determine the wet/dry ratio by dividing the net wet weight by the net dry weight:
[Wet Sediment Weight (g)]/[Dry Sediment Weight (g)] = Wet to Dry Ratio [1]2. Determine the density (g/mL) of the wet control or dilution sediment. This shall be used to determine total volume of wet sediment needed for the various test treatments.
[Mean Wet Sediment Weight (g)]/[Mean Wet Sediment Volume (mL)] = Wet Sediment Density (g/mL) [2]3. To determine the amount of base fluid needed to obtain a test concentration of 500 mg base fluid per kg dry sediment use the following formulas:
Determine the amount of wet sediment required:
[Wet Sediment Density (g/mL)] × [Volume of Sediment Required per Concentration (mL)] = Weight Wet Sediment Required per Conc. (g) [3]Determine the amount of dry sediment in kilograms (kg) required for each concentration:
{[Wet Sediment per Concentration (g)]/[Mean Wet to Dry Ratio]} × (1kg/1000g) = Dry Weight Sediment (kg) [4]Finally, determine the amount of base fluid required to spike the control sediment at each concentration:
[Conc. Desired (mg/kg)] × [Dry Weight Sediment (kg)] = Base Fluid Required (mg) [5]For spiking test substances other than pure base fluids (e.g., whole mud formulations), determine the spike amount as follows:
[Conc. Desired (mL/kg)] × [Dry Weight Sediment (kg)] × [Test Substance Density (g/mL)] = Test Substance Required (g) [6]4. For primary mixing, place appropriate amounts of weighed base fluid into stainless mixing bowls, tare the vessel weight, then add sediment and mix with a high-shear dispersing impeller for 9 minutes. The concentration of base fluid in sediment from this mix, rather than the nominal concentration, shall be used in calculating LC5. values.
5. Tests for homogeneity of base fluid in sediment are to be performed during the procedure development phase. Because of difficulty of homogeneously mixing base fluid with sediment, it is important to demonstrate that the base fluid is evenly mixed with sediment. The sediment shall be analyzed for total petroleum hydrocarbons (TPH) using EPA Methods 3550A and 8015M, with samples taken both prior to and after distribution to replicate test containers. Base-fluid content is measured as TPH. After mixing the sediment, a minimum of three replicate sediment samples shall be taken prior to distribution into test containers. After the test sediment is distributed to test containers, an additional three sediment samples shall be taken from three test containers to ensure proper distribution of base fluid within test containers. Base-fluid content results shall be reported within 48 hours of mixing. The coefficient of variation (CV) for the replicate samples must be less than 20%. If base-fluid content results are not within the 20% CV limit, the test sediment shall be remixed. Tests shall not begin until the CV is determined to be below the maximum limit of 20%. During the test, a minimum of three replicate containers shall be sampled to determine base-fluid content during each sampling period.
6. Mix enough sediment in this way to allow for its use in the preparation of all test concentrations and as a negative control. When commencing the sediment toxicity test, range-finding tests may be required to determine the concentrations that produce a toxic effect if these data are otherwise unavailable. The definitive test shall bracket the LC5., which is the desired endpoint. The results for the base fluids shall be reported in mg of base fluid per kg of dry sediment.
ReferencesAmerican Society for Testing and Materials (ASTM). 1996. Standard Guide for Collection, Storage, Characterization, and Manipulation of Sediments for Toxicological Testing. ASTM E 1391-94. Annual Book of ASTM Standards, Volume 11.05, pp. 805-825.
Ditsworth, G.R., D.W. Schults and J.K.P. Jones. 1990. Preparation of benthic substrates for sediment toxicity testing, Environ. Toxicol. Chem. 9:1523-1529.
Suedel, B.C., J.H. Rodgers, Jr. and P.A. Clifford. 1993. Bioavailability of fluoranthene in freshwater sediment toxicity tests. Environ. Toxicol. Chem. 12:155-165.
U.S. EPA. 1994. Methods for Assessing the Toxicity of Sediment-associated Contaminants with Estuarine and Marine Amphipods. EPA/600/R-94/025. Office of Research and Development, Washington, DC.
[66 FR 6901, Jan. 22, 2001]Appendix 4 to Subpart A of Part 435 - Protocol for the Determination of Degradation of Non-Aqueous Base Fluids in a Marine Closed Bottle Biodegradation Test System: Modified ISO 11734:1995 (EPA Method 1647)
40:32.0.1.1.11.1.4.7.10 :
Appendix 4 to Subpart A of Part 435 - Protocol for the Determination of Degradation of Non-Aqueous Base Fluids in a Marine Closed Bottle Biodegradation Test System: Modified ISO 11734:1995 (EPA Method 1647) 1.0. Summary of EPA Method 1647a. This method determines the anaerobic degradation potential of mineral oils, paraffin oils and non-aqueous fluids (NAF) in sediments. These substrates are base fluids for formulating offshore drilling fluids. The test evaluates base fluid biodegradation rates by monitoring gas production due to microbial degradation of the test fluid in natural marine sediment.
b. The test procedure places a mixture of marine/estuarine sediment, test substrate (hydrocarbon or controls) and seawater into clean 120 mL (150 mL actual volume) Wheaton serum bottles. The test is run using four replicate serum bottles containing 2,000 mg carbon/kg dry weight concentration of test substrate in sediment. The use of resazurin dye solution (1 ppm) evaluates the anaerobic (redox) condition of the bottles (dye is blue when oxygen is present, reddish in low oxygen conditions and colorless if oxygen free). After capping the bottles, a nitrogen sparge removes air in the headspace before incubation begins. During the incubation period, the sample should be kept at a constant temperature of 29 ±1 °C. Gas production and composition is measured approximately every two weeks. The samples need to be brought to ambient temperature before making the measurements. Measure gas production using a pressure gauge. Barometric pressure is measured at the time of testing to make necessary volume adjustments.
c. ISO 11734:1995 specifies that total gas is the standard measure of biodegradation. While modifying this test for evaluating biodegradation of NAFs, methane was also monitored and found to be an acceptable method of evaluating biodegradation. Section 7 contains the procedures used to follow biodegradation by methane production. Measurement of either total gas or methane production is permitted. If methane is followed, determine the composition of the gas by using gas chromatography (GC) analysis at each sampling. At the end of the test when gas production stops, or at around 275 days, an analysis of sediment for substrate content is possible. Common methods which have been successfully used for analyzing NAFs from sediments are listed in Section 8.
2.0 System RequirementsThis environmental test system has three phases, spiked sediment, overlying seawater, and a gas headspace. The sediment/test compound mixture is combined with synthetic sea water and transferred into 120-mL serum bottles. The total volume of sediment/sea water mixture in the bottles is 75 mL. The volume of the sediment layer will be approximately 50 mL, but the exact volume of the sediment will depend on sediment characteristics (wet:dry ratio and density). The amount of synthetic sea water will be calculated to bring the total volume in the bottles to 75 mL. The test systems are maintained at a temperature of 29 ±1 °C during incubation. The test systems are brought to ambient temperatures prior to measuring pressure or gas volume.
2.1 Sample Requirementsa. The concentration of base fluids are at least 2,000 mg carbon test material/kg dry sediment. Carbon concentration is determined by theoretical composition based on the chemical formula or by chemical analysis by ASTM D5291-96. Sediments with positive, intermediate and negative control substances as well as a C16-C18 internal olefin type base fluid will be run in conjunction with test materials under the same conditions. The positive control is ethyl oleate (CAS 111-62-6), the intermediate control is 1-hexadecene (CAS 629-73-2), and the negative control is squalane (CAS 111-01-3). Controls must be of analytical grade or the highest grade available. Each test control concentration should be prepared according to the mixing procedure described in Section 3.1.
b. Product names will be used for examples or clarification in the following text. Any use of trade or product names in this publication is for descriptive use only, and does not constitute endorsement by EPA or the authors.
2.2. Seawater RequirementsSynthetic seawater at a salinity of 25 ±1 ppt should be used for the test. The synthetic seawater should be prepared by mixing a commercially available artificial seawater mix, into high purity distilled or de-ionized water. The seawater should be aerated and allowed to age for approximately one month prior to use.
2.3. Sediment Requirementsa. The dilution sediment must be from a natural estuarine or marine environment and be free of the compounds of interest. The collection location, date and time will be documented and reported. The sediment is prepared by press-sieving through a 2,000-micron mesh sieve to remove large debris, then press-sieving through a 500-micron sieve to remove indigenous organisms that may confound test results. The water content of the sediment should be less than 60% (w/w) or a wet to dry ratio of 2.5. The sediment should have a minimum organic matter content of 3% (w/w) as determined by ASTM D2974-07a (Method A and D and calculate organic matter as in Section 8.3 of method ASTM D2974-07a).
b. To reduce the osmotic shock to the microorganisms in the sediment the salinity of the sediment's pore water should be between 20-30 ppt. Sediment should be used for testing as soon as possible after field collection. If required, sediment can be stored in the dark at 4 °C with 3-6 inches of overlying water in a sealed container for a maximum period of 2 months prior to use.
3.0 Test Set UpThe test is set up by first mixing the test or control substrates into the sediment inoculum, then mixing in seawater to make a pourable slurry. The slurry is then poured into serum bottles, which are then flushed with nitrogen and sealed.
3.1. Mixing ProcedureBecause base fluids are strongly hydrophobic and do not readily mix with sediments, care must be taken to ensure base fluids are thoroughly homogenized within the sediment. All concentrations are weight-to-weight comparisons (mg of base fluid to kg of dry control sediment). Sediment and base fluid mixing will be accomplished by using the following method.
3.1.1. Determine the wet to dry weight ratio for the control sediment by weighing approximately 10 sub-samples of approximately 1 g each of the screened and homogenized wet sediment into tared aluminum weigh pans. Dry sediment at 105 °C for 18-24 h. Remove the dried sediments and cool in a desiccator. Repeat the drying, cooling, and weighing cycle until a constant weight is achieved (within 4% of previous weight). Re-weigh the samples to determine the dry weight. Calculate the mean wet and dry weights of the 10 sub samples and determine the wet/dry ratio by dividing the mean wet weight by the mean dry weight using Equation 5-1. This is required to determine the weight of wet sediment needed to prepare the test samples.
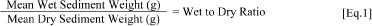
3.1.2. Determine the density (g/ml) of the wet sediment. This will be used to determine total volume of wet sediment needed for the various test treatments. One method is to tare a 5 ml graduated cylinder and add about 5 ml of homogenized sediment. Carefully record the volume then weigh this volume of sediment. Repeat this a total of three times. To determine the wet sediment density, divide the weight by volume per the following formula:
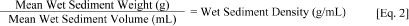
3.1.3. Determine the amount of base fluid to be spiked into wet sediment in order to obtain the desired initial base fluid concentration of 2,000 mg carbon/kg dry weight. An amount of wet sediment that is the equivalent of 30 g of dry sediment will be added to each bottle. A typical procedure is to prepare enough sediment for 8 serum bottles (3 bottles to be sacrificed at the start of the test, 4 bottles incubated for headspace analysis, and enough extra sediment for 2 extra bottles). Extra sediment is needed because some of the sediment will remain coated onto the mixing bowl and utensils. Experience with this test may indicate that preparing larger volumes of spiked sediment is a useful practice, then the following calculations should be adjusted accordingly.
a. Determine the total weight of dry sediment needed to add 30 g dry sediment to 8 bottles. If more bottles are used then the calculations should be modified accordingly. For example:
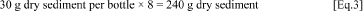
b. Determine the weight of base fluid, in terms of carbon, needed to obtain a final base fluid concentration of 2,000 mg carbon/kg dry weight. For example:
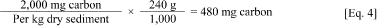
c. i. Convert from mg of carbon to mg of base fluid. This calculation will depend on the % fraction of carbon present in the molecular structure of each base fluid. For the control fluids, ethyl oleate is composed of 77.3% carbon, hexadecene is composed of 85.7% carbon, and squalane is composed of 85.3% carbon. The carbon fraction of each base fluid should be supplied by the manufacturer or determined before use. ASTM D5291-96 or equivalent will be used to determine composition of fluid.
ii. To calculate the amount of base fluid to add to the sediment, divide the amount of carbon (480 mg) by the percent fraction of carbon in the fluid.
iii. For example, the amount of ethyl oleate added to 240 g dry weight sediment can be calculated from the following equation:
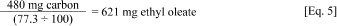
iv. Therefore, add 621 mg of ethyl oleate to 240 g dry weight sediment for a final concentration of 2,000 mg carbon/kg sediment dry weight.
3.1.4. Mix the calculated amount of base fluid with the appropriate weight of wet sediment.
a. Use the wet:dry ratio to convert from g sediment dry weight to g sediment wet weight, as follows:
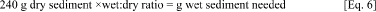
b. i. Weigh the appropriate amount of base fluid (calculated in Section 3.1.3.c) into stainless mixing bowls, tare the vessel weight, then add the wet sediment calculated in Equation 5, and mix with a high shear dispersing impeller for 9 minutes.
ii. The sediment is now mixed with synthetic sea water to form a slurry that will be transferred into the bottles.
3.2. Creating Seawater/Sediment Slurry
Given that the total volume of sediment/sea water slurry in each bottle is to be 75 mL, determine the volume of sea water to add to the wet sediment.
3.2.1. If each bottle is to contain 30 g dry sediment, calculate the weight, and then the volume, of wet sediment to be added to each bottle.

3.2.4. Convert the wet sediment weight from Equation 6 into a volume using the sediment density.
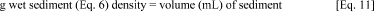
3.2.5. Determine the amount of sea water to mix with the wet sediment.
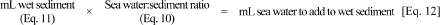
Mix sea water thoroughly with wet sediment to form a sediment/sea water slurry.
3.3. Bottling the Sediment Seawater Slurry
The total volume of sediment/sea water slurry in each bottle is to be 75 mL. Convert the volume (mL) of sediment/sea water slurry into a weight (g) using the density of the sediment and the seawater.

This should provide each bottle with 30 g dry sediment in a total volume of 75 mL.
3.3.4. Putting the sediment:seawater slurry in the serum bottles.
a. Note: The slurry will need to be constantly stirred to keep the sediment suspended.
b. Place a tared serum bottle on a balance and add the appropriate amount of slurry to the bottle using a funnel. Once the required slurry is in the bottle remove the funnel, add 2-3 drops (25 µL) of a 1 gram/L resazurin dye stock solution. Cap the bottle with a butyl rubber stopper (Bellco Glass, Part #2048-11800) and crimp with an aluminum seal (Bellco Glass Part #2048-11020).
c. Using a plastic tube with a (23-gauge, 1-inch long) needle attached to one side and a nitrogen source to the other, puncture the serum cap with the needle. Puncture the serum cap again with a second needle to sparge the bottle's headspace of residual air for two minutes. The nitrogen should be flowing at no more than 100 mL/min to encourage gentle displacement of oxygenated air with nitrogen. Faster nitrogen flow rates would cause mixing and complete oxygen removal would take much longer. Remove the nitrogen needle first to avoid any initial pressure problems. The second (vent) needle should be removed within 30 seconds of removing the nitrogen needle.
d. Triplicate blank test systems are prepared, with similar quantities of sediment and seawater without any base fluid. Incubate in the dark at a constant temperature of 29 ±1 °C.
e. Record the test temperature. The test duration is dependent on base fluid performance, but at a maximum should be no more than 275 days. Stop the test after all base fluids have achieved a plateau of gas production. At termination, base fluid concentrations can be verified in the terminated samples by extraction and GC analysis according to Section 8.
4.0. Concentration Verification Chemical Analysesa. Because of the difficulty of homogeneously mixing base fluid with sediment, it is important to demonstrate that the base fluid is evenly mixed within the sediment sea water slurry that was added to each bottle. Of the seven serum bottles set up for each test or control condition, three are randomly selected for concentration verification analyses. These should be immediately placed at 4 °C and a sample of sediment from each bottle should be analyzed for base fluid content as soon as possible. The coefficient of variation (CV) for the replicate samples must be less than 20%. The results should show recovery of at least 70% of the spiked base fluid. Use an appropriate analytical procedure described in Section 8 to perform the extractions and analyses. If any set of sediments fail the criteria for concentration verification, then the corrective action for that set of sediments is also outlined in Section 8.
b. The nominal concentrations and the measured concentrations from the three bottles selected for concentration verification should be reported for the initial test concentrations. The coefficient of variation (CV) for the replicate samples must be less than 20%. If base fluid content results are not within the 20% CV limit, the test must be stopped and restarted with adequately mixed sediment.
5.0. Gas Monitoring ProceduresBiodegradation is measured by total gas as specified in ISO 11734:1995. Methane production can also be tracked and is described in Section 7.
5.1. Total Gas Monitoring ProceduresBottles should be brought to room temperature before readings are taken. a. The bottles are observed to confirm that the resazurin has not oxidized to pink or blue. Total gas production in the culture bottles should be measured using a pressure transducer (one source is Biotech International). The pressure readings from test and control cultures are evaluated against a calibration curve created by analyzing the pressure created by known additions of gas to bottles established identically to the culture bottles. Bottles used for the standard curve contain 75 mL of water, and are sealed with the same rubber septa and crimp cap seals used for the bottles containing sediment. After the bottles used in the standard curve have been sealed, a syringe needle inserted through the septa is used to equilibrate the pressure inside the bottles to the outside atmosphere. The syringe needle is removed and known volumes of air are injected into the headspace of the bottles. Pressure readings provide a standard curve relating the volume of gas injected into the bottles and headspace pressure. No less than three points may be used to generate the standard curve. A typical standard curve may use 0, 1, 5, 10, 20 and 40 mL of gas added to the standard curve bottles.
b. The room temperature and barometric pressure (to two digits) should be recorded at the time of sampling. One option for the barometer is Fisher Part #02-400 or 02-401. Gas production by the sediment is expressed in terms of the volume (mL) of gas at standard temperature (0 °C = 273 °K) and pressure (1 atm = 30 inches of Hg) using Eq. 16.
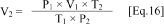 Where: V2
= Volume of gas production at standard temperature and pressure P1
= Barometric pressure on day of sampling (inches of Hg) V1 = Volume
of gas measured on day of sampling (mL) T2 = Standard temperature =
273 °K T1 = Temperature on day of sampling ( °C + 273 = °K) P2 =
Standard pressure = 30 inches Hg
Where: V2
= Volume of gas production at standard temperature and pressure P1
= Barometric pressure on day of sampling (inches of Hg) V1 = Volume
of gas measured on day of sampling (mL) T2 = Standard temperature =
273 °K T1 = Temperature on day of sampling ( °C + 273 = °K) P2 =
Standard pressure = 30 inches Hg
c. An estimate can be made of the total volume of anaerobic gas that will be produced in the bottles. The gas production measured for each base fluid can be expressed as a percent of predicted total anaerobic gas production.
5.1.1. Calculate the total amount of carbon in the form of the base fluid present in each bottle.
a. Each bottle is to contain 30 g dry weight sediment. The base fluid concentration is 2,000 mg carbon/kg dry weight sediment. Therefore:
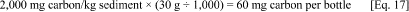
5.1.2. Theory states that anaerobic microorganisms will convert 1 mole of carbon substrate into 1 mole of total anaerobic gas production.
a. Calculate the number of moles of carbon in each bottle.
b. The molecular weight of carbon is 12 (i.e., 1 mole of carbon = 12 g). Therefore, the number of moles of carbon in each bottle can be calculated.
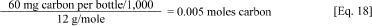
5.1.3. Calculate the predicted volume of anaerobic gas.
One mole of gas equals 22.4 L (at standard temperature and pressure), therefore,
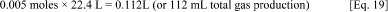 5.2. Gas
Venting
5.2. Gas
Venting
a. If the pressure in the serum bottle is too great for the pressure transducer or syringe, some of the excess gas must be wasted. The best method to do this is to vent the excess gas right after measurement. To do this, remove the barrel from a 10-mL syringe and fill it 1/3 full with water. This is then inserted into the bottle through the stopper using a small diameter (high gauge) needle. The excess pressure is allowed to vent through the water until the bubbles stop. This allows equalization of the pressure inside the bottle to atmospheric without introducing oxygen. The amount of gas vented (which is equal to the volume determined that day) must be kept track of each time the bottles are vented. A simple way to do this in a spreadsheet format is to have a separate column in which cumulative vented gas is tabulated. Each time the volume of gas in the cultures is analyzed, the total gas produced is equal to the gas in the culture at that time plus the total of the vented gas.
b. To keep track of the methane lost in the venting procedure, multiply the amount of gas vented each time by the corrected % methane determined on that day. The answer gives the volume of methane wasted. This must be added into the cumulative totals similarly to the total gas additions.
6.0. Test Acceptability and Interpretation 6.1. Test AcceptabilityAt day 275 or when gas production has plateaued, whichever is first, the controls are evaluated to confirm that the test has been performed appropriately. In order for this modification of the closed bottle biodegradation test to be considered acceptable, all the controls must meet the biodegradation levels indicated in Table 1. The intermediate control hexadecene must produce at least 30% of the theoretical gas production. This level may be reexamined after two years and more data has been generated.
Table 1 - Test Acceptability Criteria
| Concentration | Percent biodegradability as a function of gas measurement | ||
|---|---|---|---|
| Positive control | Squalane negative control | Hexadecene intermediate control | |
| 2,000 mg carbon/kg | ≥60% theoretical | ≤5% theoretical | ≥30% theoretical. |
a. In order for a fluid to pass the closed bottle test, the biodegradation of the base fluid as indicated by the total amount of total gas (or methane) generated once gas production has plateaued (or at the end of 275 days, which ever is first) must be greater than or equal to the volume of gas (or methane) produced by the reference standard (internal elefin or ester).
b. The method for evaluating the data to determine whether a fluid has passed the biodegradation test must use the equations:
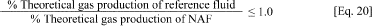 Where:
NAF = Stock base fluid being tested for compliance Reference fluid
= C16-C18 internal olefin or C12 -C14 or C8 ester reference fluid
7.0. Methane Measurement 7.1. Methane Monitoring Procedures
Where:
NAF = Stock base fluid being tested for compliance Reference fluid
= C16-C18 internal olefin or C12 -C14 or C8 ester reference fluid
7.0. Methane Measurement 7.1. Methane Monitoring Procedures
a. The use of total gas production alone may result in an underestimation of the actual metabolism occurring since CO2 is slightly soluble in water. An acceptable alternative method is to monitor methane production and total gas production. This is easily done using GC analysis. A direct injection of headspace gases can be made into a GC using almost any packed or capillary column with an FID detector. Unless volatile fuels or solvents are present in the test material or the inocula, the only component of the headspace gas that can be detected using an FID detector is methane. The percent methane in the headspace gas is determined by comparing the response of the sample injections to the response from injections of known percent methane standards. The percent methane is corrected for water vapor saturation using Eq. 21 and then converted to a volume of dry methane using Eq. 22.
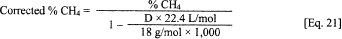 Where: D
= The density of water vapor at saturation (g/m 3, can be found in
CRC Handbook of Chemistry and Physics) for the temperature of
sampling.
Where: D
= The density of water vapor at saturation (g/m 3, can be found in
CRC Handbook of Chemistry and Physics) for the temperature of
sampling. 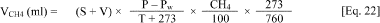 Where: VCH4 = Volume of methane in the bottle S = Volume of excess
gas production (measured with a pressure transducer) V = Volume of
the headspace in the culture bottle (total volume - liquid phase) P
= Barometric pressure (mm Hg, measured with barometer) T =
Temperature (°C) Pw = Vapor pressure of water at T (mm Hg, can be
found in CRC Handbook of Chemistry and Physics) CH4 = % methane in
headspace gas (after correction for water vapor)
Where: VCH4 = Volume of methane in the bottle S = Volume of excess
gas production (measured with a pressure transducer) V = Volume of
the headspace in the culture bottle (total volume - liquid phase) P
= Barometric pressure (mm Hg, measured with barometer) T =
Temperature (°C) Pw = Vapor pressure of water at T (mm Hg, can be
found in CRC Handbook of Chemistry and Physics) CH4 = % methane in
headspace gas (after correction for water vapor)
b. The total volume of serum bottles sold as 125 mL bottles (Wheaton) is 154.8 mL.
c. The volumes of methane produced are then compared to the volumes of methane in the controls to determine if a significant inhibition of methane production or a significant increase of methane production has been observed. Effective statistical analyses are important, as variability in the results is common due to the heterogeneity of the inoculum's source. It is also common to observe that the timing of the initiation of culture activity is not equal in all of the cultures. Expect a great variability over the period when the cultures are active, some replicates will start sooner than others, but all of the replicates should eventually reach similar levels of base fluid degradation and methane production.
7.2. Expected Methane Production Calculationsa. The amount of methane expected can be calculated using the equation of Symons and Buswell (Eq. 23). In the case of complete mineralization, all of the carbon will appear as wither CO2 or CH4, thus the total moles of gas produced will be equal to the total moles of carbon in the parent molecule. The use of the Buswell equation allows you to calculate the effects the redox potential will have on the distribution of the products in methanogenic cultures. More reduced electron donors will allow the production of more methane, while more oxidized electron donors will cause a production of more carbon dioxide.
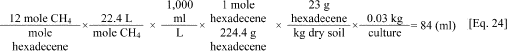
b. An example calculation of the expected methane volume in a culture fed 2,000 mg/kg hexadecene is as follows. The application of Symons and Buswell's equation reveals that hexadecene (C16H32) will yield 4 moles of CO2 and 12 moles of CH4. Assuming 30 g of dry sediment are added to the bottles with 2,334 mg hexadecene/kg dry sediment (i.e., equivalent to 2,000 mg carbon/kg dry sediment) the calculation is as follows.
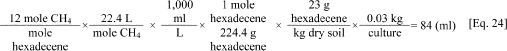
c. By subtracting the average amount of methane in control bottles from the test bottles and then dividing by the expected volume an evaluation of the completion of the process may be conducted.
8.0. Concentration Verification AnalysisThe Concentration Verification analysis is required at the beginning of the test to ensure homogeneity and confirm that the required amount of fluid was delivered to the sediments at the start of the test.
8.1. Three samples per fluid need to be analyzed and achieve ≤20% Coefficient of Variability and an average of ≥70% to ≤120% of fluid delivered to sediment.
8.2. If a third party performs the analysis, then the laboratory should be capable of delivering the homogeneity data within seven days, in order to identify any samples that do not meet the homogeneity requirement as quickly as possible.
8.3. If one sediment/fluid set, out a multiple set batch of samples, fails these criteria, then that one set of samples must be discarded and a fresh set of spiked sediment prepared, started, and analyzed to ensure homogeneity. The same stock sediment is used to prepare the replacement set(s). The remaining sets do not need to be re-mixed or restarted.
8.4. The re-mixed set(s) will need to be run the additional days as appropriate to ensure that the total number of days is the same for all sets of bottles, even though the specific days are not aligned.
8.5. Re-mixing of bottle sets can be performed multiple times as a result of a failure of the analytical criteria, until the holding time for the stock sediment has expired (60 days). If the problem set(s) has not fallen within the acceptable analytical criteria by then, it must not be part of the batch of bottles run. If the problem batch is one of the controls, and those controls were not successfully prepared when the sediment holding time expired, then the entire test must be restarted.
9.0 Program Quality Assurance and Quality Control 9.1 Calibration9.1.1. All equipment/instrumentation will be calibrated in accordance with the test method or the manufacturer's instructions and may be scheduled or triggered.
9.1.2. Where possible, standards used in calibration will be traceable to a nationally recognized standard (e.g., certified standard by NIST).
9.1.3. All calibration activities will be documented and the records retained.
9.1.4. The source, lot, batch number, and expiration date of all reagents used with be documented and retained.
9.2. Maintenance9.2.1. All equipment/instrumentation will be maintained in accordance with the test method or the manufacturer's instructions and may be scheduled or triggered.
9.2.2. All maintenance activities will be documented and the records retained.
9.3. Data Management and Handling9.3.1. All primary (raw) data will be correct, complete, without selective reporting, and will be maintained.
9.3.2. Hand-written data will be recorded in lab notebooks or electronically at the time of observation.
9.3.3. All hand-written records will be legible and amenable to reproduction by electrostatic copiers.
9.3.4. All changes to data or other records will be made by:
a. Using a single line to mark-through the erroneous entry (maintaining original data legibility).
b. Write the revision.
c. Initial, date, and provide revision code (see attached or laboratory's equivalent).
9.3.5. All data entry, transcriptions, and calculations will be verified by a qualified person.
a. Verification will be documented by initials of verifier and date.
9.3.6. Procedures will be in place to address data management procedures used (at minimum):
a. Significant figures.
b. Rounding practices.
c. Identification of outliers in data series.
d. Required statistics.
9.4. Document Control9.4.1. All technical procedures, methods, work instructions, standard operating procedures must be documented and approved by laboratory management prior to the implementation.
9.4.2. All primary data will be maintained by the contractor for a minimum of five (5) years.
9.5. Personnel and Training9.5.1. Only qualified personnel shall perform laboratory activities.
9.5.2. Records of staff training and experience will be available. This will include initial and refresher training (as appropriate).
9.6. Test Performance9.6.1. All testing will done in accordance with the specified test methods.
9.6.2. Receipt, arrival condition, storage conditions, dispersal, and accountability of the test article will be documented and maintained.
9.6.3. Receipt or production, arrival or initial condition, storage conditions, dispersal, and accountability of the test matrix (e.g., sediment or artificial seawater) will be documented and maintained.
9.6.4. Source, receipt, arrival condition, storage conditions, dispersal, and accountability of the test organisms (including inoculum) will be documented and maintained.
9.6.5. Actual concentrations administered at each treatment level will be verified by appropriate methodologies.
9.6.6. Any data originating at a different laboratory will be identified and the laboratory fully referenced in the final report.
9.7. The following references identify analytical methods that have historically been successful for achieving the analytical quality criteria.9.7.1. Continental Shelf Associates Report 1998. Joint EPA/Industry Screening Survey to Assess the Deposition of Drill Cuttings and Associated Synthetic Based Mud on the Seabed of the Louisiana Continental Shelf, Gulf of Mexico. Analysis by Charlie Henry Report Number IES/RCAT97-36 GC-FID and GC/MS.
9.7.2. EPA Method 3550 for extraction with EPA Method 8015 for GC-FID. EPA Method 3550C, Revision 3. February 2007. Ultrasonic Extraction. EPA Method 8015C, Revision 3. February 2007. Nonhalogenated Organics by Gas Chromatography.
9.7.3. Chandler, J.E., S.P. Rabke, and A.J.J. Leuterman. 1999. Predicting the Potential Impact of Synthetic-Based Muds With the Use of Biodegradation Studies. Society of Petroleum Engineers SPE 52742.
9.7.4. Chandler, J.E., B. Lee, S.P. Rabke, J.M. Geliff, R. Stauffer, and J. Hein. 2000. Modification of a Standardized Anaerobic Biodegradation Test to Discriminate Performance of Various Non-Aqueous Base Fluids. Society of Petroleum Engineers SPE 61203.
9.7.5. Munro, P.D., B Croce, C.F. Moffet, N.A Brown, A.D. McIntosh, S.J. Hird, and R.M. Stagg. 1998. Solid-Phase Test for Comparison for Degradation Rates of Synthetic Mud Base Fluids Used in the Off-shore Drilling Industry. Environ. Toxicol. Chem. 17:1951-1959.
9.7.6. Webster, L., P.R. Mackie, S.J. Hird, P.D. Munro, N.A. Brown, and C.F. Moffat. 1997. Development of Analytical Methods for the Determination of Synthetic Mud Base Fluids in Marine Sediments. The Analyst 122:1485-1490.
9.8 The following standards are approved for incorporation by reference by the Director of the Federal Register in accordance with 5 U.S.C. 552(a) and 1 CFR part 51. Copies may also be inspected at EPA's Water Docket, 1200 Pennsylvania Ave. NW., Washington, DC 20460 and at at the National Archives and Records Administration (NARA). For information on the availability of this material at NARA, call 202-741-6030, or go to: http://www.archives.gov/federal_register/code_of_federal_regulations/ibr_locations.html.
9.8.1 ASTM International. Available from ASTM International, 100 Barr Harbor Drive, P.O. Box C700, West Conshohocken, PA 19428-2959, or online at http://www.astm.org.
9.8.1.1 ASTM D5291-96, Standard Test Methods for Instrumental Determination of Carbon, Hydrogen, and Nitrogen in Petroleum Products and Lubricants, approved April 10, 1996.
9.8.1.2 ASTM D2974-07a, Standard Test Methods for Moisture, Ash, and Organic Matter of Peat and Other Organic Soils, approved March 15, 2007.
[77 FR 29837, May 18, 2012]Appendix 5 to Subpart A of Part 435 - Determination of Crude Oil Contamination in Non-Aqueous Drilling Fluids by Gas Chromatography/Mass Spectrometry (GC/MS) (EPA Method 1655)
40:32.0.1.1.11.1.4.7.11 :
Appendix 5 to Subpart A of Part 435 - Determination of Crude Oil Contamination in Non-Aqueous Drilling Fluids by Gas Chromatography/Mass Spectrometry (GC/MS) (EPA Method 1655) 1.0 Scope and Application1.1 This method determines crude (formation) oil contamination, or other petroleum oil contamination, in non-aqueous drilling fluids (NAFs) by comparing the gas chromatography/mass spectrometry (GC/MS) fingerprint scan and extracted ion scans of the test sample to that of an uncontaminated sample.
1.2 This method can be used for monitoring oil contamination of NAFs or monitoring oil contamination of the base fluid used in the NAF formulations.
1.3 Any modification of this method beyond those expressly permitted shall be considered as a major modification subject to application and approval of alternative test procedures under 40 CFR 136.4 and 136.5.
1.4 The gas chromatography/mass spectrometry portions of this method are restricted to use by, or under the supervision of analysts experienced in the use of GC/MS and in the interpretation of gas chromatograms and extracted ion scans. Each laboratory that uses this method must generate acceptable results using the procedures described in Sections 7, 9.2, and 12 of this appendix.
2.0 Summary of Method2.1 Analysis of NAF for crude oil contamination is a step-wise process. The analyst first performs a qualitative assessment of the presence or absence of crude oil in the sample. If crude oil is detected during this qualitative assessment, the analyst must perform a quantitative analysis of the crude oil concentration.
2.2 A sample of NAF is centrifuged to obtain a solids free supernate.
2.3 The test sample is prepared by removing an aliquot of the solids free supernate, spiking it with internal standard, and analyzing it using GC/MS techniques. The components are separated by the gas chromatograph and detected by the mass spectrometer.
2.4 Qualitative identification of crude oil contamination is performed by comparing the Total Ion Chromatograph (TIC) scans and Extracted Ion Profile (EIP) scans of test sample to that of uncontaminated base fluids, and examining the profiles for chromatographic signatures diagnostic of oil contamination.
2.5 The presence or absence of crude oil contamination observed in the full scan profiles and selected extracted ion profiles determines further sample quantitation and reporting requirements.
2.6 If crude oil is detected in the qualitative analysis, quantitative analysis must be performed by calibrating the GC/MS using a designated NAF spiked with known concentrations of a designated oil.
2.7 Quality is assured through reproducible calibration and testing of GC/MS system and through analysis of quality control samples.
3.0 Definitions3.1 A NAF is one in which the continuous - phase is a water immiscible fluid such as an oleaginous material (e.g., mineral oil, enhance mineral oil, paraffinic oil, or synthetic material such as olefins and vegetable esters).
3.2 TIC - Total Ion Chromatograph.
3.3 EIP - Extracted Ion Profile.
3.4 TCB - 1,3,5-trichlorobenzene is used as the internal standard in this method.
3.5 SPTM - System Performance Test Mix standards are used to establish retention times and monitor detection levels.
4.0 Interferences and Limitations4.1 Solvents, reagents, glassware, and other sample processing hardware may yield artifacts and/or elevated baselines causing misinterpretation of chromatograms.
4.2 All Materials used in the analysis shall be demonstrated to be free from interferences by running method blanks. Specific selection of reagents and purification of solvents by distillation in all-glass systems may be required.
4.3 Glassware shall be cleaned by rinsing with solvent and baking at 400 °C for a minimum of 1 hour.
4.4 Interferences may vary from source to source, depending on the diversity of the samples being tested.
4.5 Variations in and additions of base fluids and/or drilling fluid additives (emulsifiers, dispersants, fluid loss control agents, etc.) might also cause interferences and misinterpretation of chromatograms.
4.6 Difference in light crude oils, medium crude oils, and heavy crude oils will result in different responses and thus different interpretation of scans and calculated percentages.
5.0 Safety5.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely determined; however each chemical shall be treated as a potential health hazard. Exposure to these chemicals should be reduced to the lowest possible level.
5.2 Unknown samples may contain high concentration of volatile toxic compounds. Sample containers should be opened in a hood and handled with gloves to prevent exposure. In addition, all sample preparation should be conducted in a fume hood to limit the potential exposure to harmful contaminates.
5.3 This method does not address all safety issues associated with its use. The laboratory is responsible for maintaining a safe work environment and a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material safety data sheets (MSDSs) shall be available to all personnel involved in these analyses. Additional references to laboratory safety can be found in References 16.1 through 16.3.
5.4 NAF base fluids may cause skin irritation, protective gloves are recommended while handling these samples.
6.0 Apparatus and Materials Note:Brand names, suppliers, and part numbers are for illustrative purposes only. No endorsement is implied. Equivalent performance may be achieved using apparatus and materials other than those specified here, but demonstration of equivalent performance meeting the requirements of this method is the responsibility of the laboratory.
6.1 Equipment for glassware cleaning.
6.1.1 Laboratory sink with overhead fume hood.
6.1.2 Kiln - Capable of reaching 450 °C within 2 hours and holding 450 °C within ±10 °C, with temperature controller and safety switch (Cress Manufacturing Co., Santa Fe Springs, CA B31H or X31TS or equivalent).
6.2 Equipment for sample preparation.
6.2.1 Laboratory fume hood.
6.2.2 Analytical balance - Capable of weighing 0.1 mg.
6.2.3 Glassware.
6.2.3.1 Disposable pipettes - Pasteur, 150 mm long by 5 mm ID (Fisher Scientific 13-678-6A, or equivalent) baked at 400 °C for a minimum of 1 hour.
6.2.3.2 Glass volumetric pipettes or gas tight syringes - 1.0-mL ±1% and 0.5-mL ±1%.
6.2.3.3 Volumetric flasks - Glass, class A, 10-mL, 50-mL and 100-mL.
6.2.3.4 - Sample vials - Glass, 1- to 3-mL (baked at 400 °C for a minimum of 1 hour) with PTFE-lined screw or crimp cap.
6.2.3.5 Centrifuge and centrifuge tubes - Centrifuge capable of 10,000 rpm, or better, (International Equipment Co., IEC Centra MP4 or equivalent) and 50-mL centrifuge tubes (Nalgene, Ultratube, Thin Wall 25 × 89 mm, #3410-2539).
6.3 Gas Chromatograph/Mass Spectrometer (GC/MS):
6.3.1 Gas Chromatograph - An analytical system complete with a temperature-programmable gas chromatograph suitable for split/splitless injection and all required accessories, including syringes, analytical columns, and gases.
6.3.1.1 Column - 30 m (or 60 m) × 0.32 mm ID (or 0.25 mm ID) 1 µm film thickness (or 0.25 µm film thickness) silicone-coated fused-silica capillary column (J&W Scientific DB-5 or equivalent).
6.3.2 Mass Spectrometer - Capable of scanning from 35 to 600 amu every 1 sec or less, using 70 volts (nominal) electron energy in the electron impact ionization mode (Hewlett Packard 5970MS or comparable).
6.3.3 GC/MS interface - the interface is a capillary-direct interface from the GC to the MS.
6.3.4 - Data system - A computer system must be interfaced to the mass spectrometer. The system must allow the continuous acquisition and storage on machine-readable media of all mass spectra obtained throughout the duration of the chromatographic program. The computer must have software that can search any GC/MS data file for ions of a specific mass and that can plot such ion abundance versus retention time or scan number. This type of plot is defined as an Extracted Ion Current Profile (EIP). Software must also be available that allows integrating the abundance in any total ion chromatogram (TIC) or EIP between specified retention time or scan-number limits. It is advisable that the most recent version of the EPA/NIST Mass Spectral Library be available.
7.0 Reagents and Standards7.1 Methylene chloride - Pesticide grade or equivalent. Use when necessary for sample dilution.
7.2 Standards - Prepare from pure individual standard materials or purchase as certified solutions. If compound purity is 96% or greater, the weight may be used without correction to compute the concentration of the standard.
7.2.1 Crude Oil Reference - Obtain a sample of a crude oil with a known API gravity. This oil shall be used in the calibration procedures.
7.2.2 Synthetic Base Fluid - Obtain a sample of clean internal olefin (IO) Lab drilling fluid (as sent from the supplier - has not been circulated downhole). This drilling fluid shall be used in the calibration procedures.
7.2.3 Internal standard - Prepare a 0.01 g/mL solution of 1,3,5-trichlorobenzene (TCB). Dissolve 1.0 g of TCB in methylene chloride and dilute to volume in a 100-mL volumetric flask. Stopper, vortex, and transfer the solution to a 150-mL bottle with PTFE-lined cap. Label appropriately, and store at −5 °C to 20 °C. Mark the level of the meniscus on the bottle to detect solvent loss.
7.2.4 GC/MS system performance test mix (SPTM) standards - The SPTM standards shall contain octane, decane, dodecane, tetradecane, tetradecene, toluene, ethylbenzene, 1,2,4-trimethylbenzene, 1-methylnaphthalene and 1,3-dimethylnaphthalene. These compounds can be purchased individually or obtained as a mixture (i.e., Supelco, Catalog No. 4-7300). Prepare a high concentration of the SPTM standard at 62.5 mg/mL in methylene chloride. Prepare a medium concentration SPTM standard at 1.25 mg/mL by transferring 1.0 mL of the 62.5 mg/mL solution into a 50 mL volumetric flask and diluting to the mark with methylene chloride. Finally, prepare a low concentration SPTM standard at 0.125 mg/mL by transferring 1.0 mL of the 1.25 mg/mL solution into a 10-mL volumetric flask and diluting to the mark with methylene chloride.
7.2.5 Crude oil/drilling fluid calibration standards - Prepare a 4-point crude oil/drilling fluid calibration at concentrations of 0% (no spike - clean drilling fluid), 0.5%, 1.0%, and 2.0% by weight according to the procedures outlined in this appendix using the Reference Crude Oil:
7.2.5.1 Label 4 jars with the following identification: Jar 1 - 0%Ref-IOLab, Jar 2 - 0.5%Ref-IOLab, Jar 3 - 1%Ref-IOLab, and Jar 4 - 2%Ref-IOLab.
7.2.5.2 Weigh 4, 50-g aliquots of well mixed IO Lab drilling fluid into each of the 4 jars.
7.2.5.3 Add Reference Oil at 0.5%, 1.0%, and 2.0% by weight to jars 2, 3, and 4 respectively. Jar 1 shall not be spiked with Reference Oil in order to retain a “0%” oil concentration.
7.2.5.4 Thoroughly mix the contents of each of the 4 jars, using clean glass stirring rods.
7.2.5.5 Transfer (weigh) a 30-g aliquot from Jar 1 to a labeled centrifuge tube. Centrifuge the aliquot for a minimum of 15 min at approximately 15,000 rpm, in order to obtain a solids free supernate. Weigh 0.5 g of the supernate directly into a tared and appropriately labeled GC straight vial. Spike the 0.5-g supernate with 500 µL of the 0.01g/mL 1,3,5-trichlorobenzene internal standard solution (see Section 7.2.3 of this appendix), cap with a Teflon lined crimp cap, and vortex for ca. 10 sec.
7.2.5.6 Repeat step 7.2.5.5 except use an aliquot from Jar 2.
7.2.5.7 Repeat step 7.2.5.5 except use an aliquot from Jar 3.
7.2.5.8 Repeat step 7.2.5.5 except use an aliquot from Jar 4.
7.2.5.9 These 4 crude/oil drilling fluid calibration standards are now used for qualitative and quantitative GC/MS analysis.
7.2.6 Precision and recovery standard (mid level crude oil/drilling fluid calibration standard) - Prepare a mid point crude oil/ drilling fluid calibration using IO Lab drilling fluid and Reference Oil at a concentration of 1.0% by weight. Prepare this standard according to the procedures outlined in Section 7.2.5.1 through 7.2.5.5 of this appendix, with the exception that only “Jar 3” needs to be prepared. Remove and spike with internal standard, as many 0.5-g aliquots as needed to complete the GC/MS analysis (see Section 11.6 of this appendix - bracketing authentic samples every 12 hours with precision and recovery standard) and the initial demonstration exercise described in Section 9.2 of this appendix.
7.2.7 Stability of standards
7.2.7.1 When not used, standards shall be stored in the dark, at −5 to −20 °C in screw-capped vials with PTFE-lined lids. Place a mark on the vial at the level of the solution so that solvent loss by evaporation can be detected. Bring the vial to room temperature prior to use.
7.2.7.2 Solutions used for quantitative purposes shall be analyzed within 48 hours of preparation and on a monthly basis thereafter for signs of degradation. A standard shall remain acceptable if the peak area remains within ±15% of the area obtained in the initial analysis of the standard.
8.0 Sample Collection Preservation and Storage8.1 Collect NAF and base fluid samples in 100- to 200-mL glass bottles with PTFE- or aluminum foil lined caps.
8.2 Samples collected in the field shall be stored refrigerated until time of preparation.
8.3 Sample and extract holding times for this method have not yet been established. However, based on initial experience with the method, samples should be analyzed within seven to ten days of collection and extracts should be analyzed within seven days of preparation.
8.4 After completion of GC/MS analysis, extracts shall be refrigerated at 4 °C until further notification of sample disposal.
9.0 Quality Control9.1 Each laboratory that uses this method is required to operate a formal quality assurance program (Reference 16.4). The minimum requirements of this program shall consist of an initial demonstration of laboratory capability, and ongoing analysis of standards, and blanks as a test of continued performance, analyses of spiked samples to assess accuracy and analysis of duplicates to assess precision. Laboratory performance shall be compared to established performance criteria to determine if the results of analyses meet the performance characteristics of the method.
9.1.1 The analyst shall make an initial demonstration of the ability to generate acceptable accuracy and precision with this method. This ability shall be established as described in Section 9.2 of this appendix.
9.1.2 The analyst is permitted to modify this method to improve separations or lower the cost of measurements, provided all performance requirements are met. Each time a modification is made to the method, the analyst is required to repeat the calibration (Section 10.4 of this appendix) and to repeat the initial demonstration procedure described in Section 9.2 of this appendix.
9.1.3 Analyses of blanks are required to demonstrate freedom from contamination. The procedures and criteria for analysis of a blank are described in Section 9.3 of this appendix.
9.1.4 Analysis of a matrix spike sample is required to demonstrate method accuracy. The procedure and QC criteria for spiking are described in Section 9.4 of this appendix.
9.1.5 Analysis of a duplicate field sample is required to demonstrate method precision. The procedure and QC criteria for duplicates are described in Section 9.5 of this appendix.
9.1.6 Analysis of a sample of the clean NAF(s) (as sent from the supplier - i.e., has not been circulated downhole) used in the drilling operations is required.
9.1.7 The laboratory shall, on an ongoing basis, demonstrate through calibration verification and the analysis of the precision and recovery standard (Section 7.2.6 of this appendix) that the analysis system is in control. These procedures are described in Section 11.6 of this appendix.
9.1.8 The laboratory shall maintain records to define the quality of data that is generated.
9.2 Initial precision and accuracy - The initial precision and recovery test shall be performed using the precision and recovery standard (1% by weight Reference Oil in IO Lab drilling fluid). The laboratory shall generate acceptable precision and recovery by performing the following operations.
9.2.1 Prepare four separate aliquots of the precision and recovery standard using the procedure outlined in Section 7.2.6 of this appendix. Analyze these aliquots using the procedures outlined in Section 11 of this appendix.
9.2.2 Using the results of the set of four analyses, compute the average recovery (X) in weight percent and the standard deviation of the recovery(s) for each sample.
9.2.3 If s and X meet the acceptance criteria of 80% to 110%, system performance is acceptable and analysis of samples may begin. If, however, s exceeds the precision limit or X falls outside the range for accuracy, system performance is unacceptable. In this event, review this method, correct the problem, and repeat the test.
9.2.4 Accuracy and precision - The average percent recovery (P) and the standard deviation of the percent recovery (Sp) Express the accuracy assessment as a percent recovery interval from P-2Sp to P + 2Sp. For example, if P = 90% and Sp = 10% for four analyses of crude oil in NAF, the accuracy interval is expressed as 70% to 110%. Update the accuracy assessment on a regular basis.
9.3 Blanks - Rinse glassware and centrifuge tubes used in the method with 30 mL of methylene chloride, remove a 0.5-g aliquot of the solvent, spike it with the 500 µL of the internal standard solution (Section 7.2.3 of this appendix) and analyze a 1-µL aliquot of the blank sample using the procedure in Section 11 of this appendix. Compute results per Section 12 of this appendix.
9.4 Matrix spike sample - Prepare a matrix spike sample according to procedure outlined in Section 7.2.6 of this appendix. Analyze the sample and calculate the concentration (% oil) in the drilling fluid and % recovery of oil from the spiked drilling fluid using the methods described in Sections 11 and 12 of this appendix.
9.5 Duplicates - A duplicate field sample shall be prepared and analyzed according to Section 11. The relative percent difference (RPD) of the calculated concentrations shall be less than 15%.
9.5.1 Analyze each of the duplicates per the procedure in Section 11 of this appendix and compute the results per Section 12 of this appendix.
9.5.2 Calculate the relative percent difference (RPD) between the two results per the following equation:
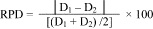 where: D1
= Concentration of crude oil in the sample; and D2 = Concentration
of crude oil in the duplicate sample.
where: D1
= Concentration of crude oil in the sample; and D2 = Concentration
of crude oil in the duplicate sample.
9.5.3 If the RPD criteria are not met, the analytical system shall be judged to be out of control, and the problem must be immediately identified and corrected, and the sample batch re-analyzed.
9.6 A clean NAF sample shall be prepared and analyzed according to Section 11. Ultimately the oil-equivalent concentration from the TIC or EIP signal measured in the clean NAF sample shall be subtracted from the corresponding authentic field samples in order to calculate the true contaminant concentration (% oil) in the field samples (see Section 12).
9.7 The specifications contained in this method can be met if the apparatus used is calibrated properly, and maintained in a calibrated state. The standards used for initial precision and recovery (Section 9.2 of this appendix) and ongoing precision and recovery (Section 11.6 of this appendix) shall be identical, so that the most precise results will be obtained. The GC/MS instrument will provide the most reproducible results if dedicated to the setting and conditions required for the analyses given in this method.
9.8 Depending on specific program requirements, field replicates and field spikes of crude oil into samples may be required when this method is used to assess the precision and accuracy of the sampling and sample transporting techniques.
10.0 Calibration10.1 Establish gas chromatographic/mass spectrometer operating conditions given in Table 1 of this appendix. Perform the GC/MS system hardware-tune as outlined by the manufacture. The gas chromatograph shall be calibrated using the internal standard technique.
Note:Because each GC is slightly different, it may be necessary to adjust the operating conditions (carrier gas flow rate and column temperature and temperature program) slightly until the retention times in Table 2 of this appendix are met.
Table 1 - Gas Chromatograph/Mass Spectrometer (GC/MS) Operation Conditions
| Parameter | Setting |
|---|---|
| Injection pot | 280 °C |
| Transfer line | 280 °C |
| Detector | 280 °C |
| Initial Temperature | 50 °C |
| Initial Time | 5 minutes |
| Ramp | 50 to 300 °C @ 5 °C per minute |
| Final Temperature | 300 °C |
| Final Hold | 20 minutes or until all peaks have eluted |
| Carrier Gas | Helium |
| Flow rate | As required for standard operation |
| Split ratio | As required to meet performance criteria (∼1:100) |
| Mass range | 35 to 600 amu |
Table 2 - Approximate Retention Time for Compounds
| Compound | Approximate retention time (minutes) |
|---|---|
| Toluene | 5.6 |
| Octane, n−C8 | 7.2 |
| Ethylbenzene | 10.3 |
| 1,2,4-Trimethylbenzene | 16.0 |
| Decane, −C10 | 16.1 |
| TCB (Internal Standard) | 21.3 |
| Dodecane, −C12 | 22.9 |
| 1-Methylnaphthalene | 26.7 |
| 1-Tetradecene | 28.4 |
| Tetradecane, −C14 | 28.7 |
| 1,3-Dimethylnaphthalene | 29.7 |
10.2 Internal standard calibration procedure - 1,3,5-trichlorobenzene (TCB) has been shown to be free of interferences from diesel and crude oils and is a suitable internal standard.
10.3 The system performance test mix standards prepared in Section 7.2.4 of this appendix shall be used to establish retention times and establish qualitative detection limits.
10.3.1 Spike a 500-mL aliquot of the 1.25 mg/mL SPTM standard with 500 µL of the TCB internal standard solution.
10.3.2 Inject 1.0 µL of this spiked SPTM standard onto the GC/MS in order to demonstrate proper retention times. For the GC/MS used in the development of this method, the ten compounds in the mixture had typical retention times shown in Table 2 of this appendix. Extracted ion scans for m/z 91 and 105 showed a maximum abundance of 400,000.
10.3.3 Spike a 500-mL aliquot of the 0.125 mg/mL SPTM standard with 500 µL of the TCB internal standard solution.
10.3.4 Inject 1.0 µL of this spiked SPTM standard onto the GC/MS to monitor detectable levels. For the GC/MS used in the development of this test, all ten compounds showed a minimum peak height of three times signal to noise. Extracted ion scans for m/z 91 and 105 showed a maximum abundance of 40,000.
10.4 GC/MS crude oil/drilling fluid calibration - There are two methods of quantification: Total Area Integration (C8-C13) and EIP Area Integration using m/z's 91 and 105. The Total Area Integration method should be used as the primary technique for quantifying crude oil in NAFs. The EIP Area Integration method should be used as a confirmatory technique for NAFs. However, the EIP Area Integration method shall be used as the primary method for quantifying oil in enhanced mineral oil (EMO) based drilling fluid. Inject 1.0 µL of each of the four crude oil/drilling fluid calibration standards prepared in Section 7.2.5 of this appendix into the GC/MS. The internal standard should elute approximately 21-22 minutes after injection. For the GC/MS used in the development of this method, the internal standard peak was (35 to 40)% of full scale at an abundance of about 3.5e + 07.
10.4.1 Total Area Integration Method - For each of the four calibration standards obtain the following: Using a straight baseline integration technique, obtain the total ion chromatogram (TIC) area from C8 to C13. Obtain the TIC area of the internal standard (TCB). Subtract the TCB area from the C8-C13 area to obtain the true C8-C13 area. Using the C8-C13 and TCB areas, and known internal standard concentration, generate a linear regression calibration using the internal standard method. The r 2 value for the linear regression curve shall be greater than or equal to 0.998. Some synthetic fluids might have peaks that elute in the window and would interfere with the analysis. In this case the integration window can be shifted to other areas of scan where there are no interfering peaks from the synthetic base fluid.
10.4.2 EIP Area Integration - For each of the four calibration standards generate Extracted Ion Profiles (EIPs) for m/z 91 and 105. Using straight baseline integration techniques, obtain the following EIP areas:
10.4.2.1 For m/z 91 integrate the area under the curve from approximately 9 minutes to 21-22 minutes, just prior to but not including the internal standard.
10.4.2.2 For m/z 105 integrate the area under the curve from approximately 10.5 minutes to 26.5 minutes.
10.4.2.3 Obtain the internal standard area from the TCB in each of the four calibration standards, using m/z 180.
10.4.2.4 Using the EIP areas for TCB, m/z 91 and m/z105, and the known concentration of internal standard, generate linear regression calibration curves for the target ions 91 and 105 using the internal standard method. The r 2 value for each of the EIP linear regression curves shall be greater than or equal to 0.998.
10.4.2.5 Some base fluids might produce a background level that would show up on the extracted ion profiles, but there should not be any real peaks (signal to noise ratio of 1:3) from the clean base fluids.
11.0 Procedure11.1 Sample Preparation -
11.1.1 Mix the authentic field sample (drilling fluid) well. Transfer (weigh) a 30-g aliquot of the sample to a labeled centrifuge tube.
11.1.2 Centrifuge the aliquot for a minimum of 15 min at approximately 15,000 rpm, in order to obtain a solids free supernate.
11.1.3 Weigh 0.5 g of the supernate directly into a tared and appropriately labeled GC straight vial.
11.1.4 Spike the 0.5-g supernate with 500 µL of the 0.01g/mL 1,3,5-trichlorobenzene internal standard solution (see Section 7.2.3 of this appendix), cap with a Teflon lined crimp cap, and vortex for ca. 10 sec.
11.1.5 The sample is ready for GC/MS analysis.
11.2 Gas Chromatography.
Table 1 of this appendix summarizes the recommended operating conditions for the GC/MS. Retention times for the n-alkanes obtained under these conditions are given in Table 2 of this appendix. Other columns, chromatographic conditions, or detectors may be used if initial precision and accuracy requirements (Section 9.2 of this appendix) are met. The system shall be calibrated according to the procedures outlined in Section 10 of this appendix, and verified every 12 hours according to Section 11.6 of this appendix.
11.2.1 Samples shall be prepared (extracted) in a batch of no more than 20 samples. The batch shall consist of 20 authentic samples, 1 blank (Section 9.3 of this appendix), 1 matrix spike sample (9.4), and 1 duplicate field sample (9.5), and a prepared sample of the corresponding clean NAF used in the drilling process.
11.2.2 An analytical sequence shall be analyzed on the GC/MS where the 3 SPTM standards (Section 7.2.4 of this appendix) containing internal standard are analyzed first, followed by analysis of the four GC/MS crude oil/drilling fluid calibration standards (Section 7.2.5 of this appendix), analysis of the blank, matrix spike sample, the duplicate sample, the clean NAF sample, followed by the authentic samples.
11.2.3 Samples requiring dilution due to excessive signal shall be diluted using methylene chloride.
11.2.4 Inject 1.0 µL of the test sample or standard into the GC, using the conditions in Table 1 of this appendix.
11.2.5 Begin data collection and the temperature program at the time of injection.
11.2.6 Obtain a TIC and EIP fingerprint scans of the sample (Table 3 of this appendix).
11.2.7 If the area of the C8 to C13 peaks exceeds the calibration range of the system, dilute a fresh aliquot of the test sample weighing 0.50-g and re-analyze.
11.2.8 Determine the C8 to C13 TIC area, the TCB internal standard area, and the areas for the m/z 91 and 105 EIPs. These shall be used in the calculation of oil concentration in the samples (see Section 12 of this appendix).
Table 3 - Recommended Ion Mass Numbers
| Selected ion mass numbers | Corresponding aromatic compounds | Typical rentention time (minutes) |
|---|---|---|
| 91 | Methylbenzene | 6.0 |
| Ethylbenzene | 10.3 | |
| 1,4-Dimethylbenzene | 10.9 | |
| 1,3-Dimethylbenzene | 10.9 | |
| 1,2-Dimethylbenzene | 11.9 | |
| 105 | 1,3,5-Trimethylbenzene | 15.1 |
| 1,2,4-Trimethylbenzene | 16.0 | |
| 1,2,3-Trimethylbenzene | 17.4 | |
| 156 | 2,6-Dimethylnaphthalene | 28.9 |
| 1,2-Dimethylnaphthalene | 29.4 | |
| 1,3-Dimethylnaphthalene | 29.7 |
11.2.9 Observe the presence of peaks in the EIPs that would confirm the presence of any target aromatic compounds. Using the EIP areas and EIP linear regression calibrations compare the abundance of the aromatic peaks, and if appropriate, determine approximate crude oil contamination in the sample for each of the target ions.
11.3 Qualitative Identification - See Section 17 of this method for schematic flowchart.
11.3.1 Qualitative identification shall be accomplished by comparison of the TIC and EIP area data from an authentic sample to the TIC and EIP area data from the calibration standards (see Section 10.4). Crude oil shall be identified by the presence of C10 to C13 n-alkanes and corresponding target aromatics.
11.3.2 Using the calibration data, establish the identity of the C8 to C13 peaks in the chromatogram of the sample. Using the calibration data, establish the identity of any target aromatics present on the extracted ion scans.
11.3.3 Crude oil is not present in a detectable amount in the sample if there are no target aromatics seen on the extracted ion scans. The experience of the analyst shall weigh heavily in the determination of the presence of peaks at a signal-to-noise ratio of 3 or greater.
11.3.4 If the chromatogram shows n-alkanes from C8 to C13 and target aromatics to be present, contamination by crude oil or diesel shall be suspected and quantitative analysis shall be determined. If there are no n-alkanes present that are not seen on the blank, and no target aromatics are seen, the sample can be considered to be free of contamination.
11.4 Quantitative Identification -
11.4.1 Determine the area of the peaks from C8 to C13 as outlined in the calibration section (10.4.1 of this appendix). If the area of the peaks for the sample is greater than that for the clean NAF (base fluid) use the crude oil/drilling fluid calibration TIC linear regression curve to determine approximate crude oil contamination.
11.4.2 Using the EIPs outlined in Section 10.4.2 of this appendix, determine the presence of any target aromatics. Using the integration techniques outlined in Section 10.4.2 of this appendix, obtain the EIP areas for m/z 91 and 105. Use the crude oil/drilling fluid calibration EIP linear regression curves to determine approximate crude oil contamination.
11.5 Complex Samples -
11.5.1 The most common interferences in the determination of crude oil can be from mineral oil, diesel oil, and proprietary additives in drilling fluids.
11.5.2 Mineral oil can typically be identified by its lower target aromatic content, and narrow range of strong peaks.
11.5.3 Diesel oil can typically be identified by low amounts of n-alkanes from C7 to C9, and the absence of n-alkanes greater than C25.
11.5.4 Crude oils can usually be distinguished by the presence of high aromatics, increased intensities of C8 to C13 peaks, and/ or the presence of higher hydrocarbons of C25 and greater (which may be difficult to see in some synthetic fluids at low contamination levels).
11.5.4.1 Oil condensates from gas wells are low in molecular weight and will normally produce strong chromatographic peaks in the C8-C13 range. If a sample of the gas condensate crude oil from the formation is available, the oil can be distinguished from other potential sources of contamination by using it to prepare a calibration standard.
11.5.4.2 Asphaltene crude oils with API gravity <20 may not produce chromatographic peaks strong enough to show contamination at levels of the calibration. Extracted ion peaks should be easier to see than increased intensities for the C8 to C13 peaks. If a sample of asphaltene crude from the formation is available, a calibration standard shall be prepared.
11.6 System and Laboratory Performance -
11.6.1 At the beginning of each 8-hour shift during which analyses are performed, GC crude oil/drilling fluid calibration and system performance test mixes shall be verified. For these tests, analysis of the medium-level calibration standard (1-% Reference Oil in IO Lab drilling fluid, and 1.25 mg/mL SPTM with internal standard) shall be used to verify all performance criteria. Adjustments and/or re-calibration (per Section 10 of this appendix) shall be performed until all performance criteria are met. Only after all performance criteria are met may samples and blanks be analyzed.
11.6.2 Inject 1.0 µL of the medium-level GC/MS crude oil/drilling fluid calibration standard into the GC instrument according to the procedures in Section 11.2 of this appendix. Verify that the linear regression curves for both TIC area and EIP areas are still valid using this continuing calibration standard.
11.6.3 After this analysis is complete, inject 1.0 µL of the 1.25 mg/mL SPTM (containing internal standard) into the GC instrument and verify the proper retention times are met (see Table 2 of this appendix).
11.6.4 Retention times - Retention time of the internal standard. The absolute retention time of the TCB internal standard shall be within the range 21.0 ±0.5 minutes. Relative retention times of the n-alkanes: The retention times of the n-alkanes relative to the TCB internal standard shall be similar to those given in Table 2 of this appendix.
11.6.17 Schematic Flowchart for Qualitative Identification
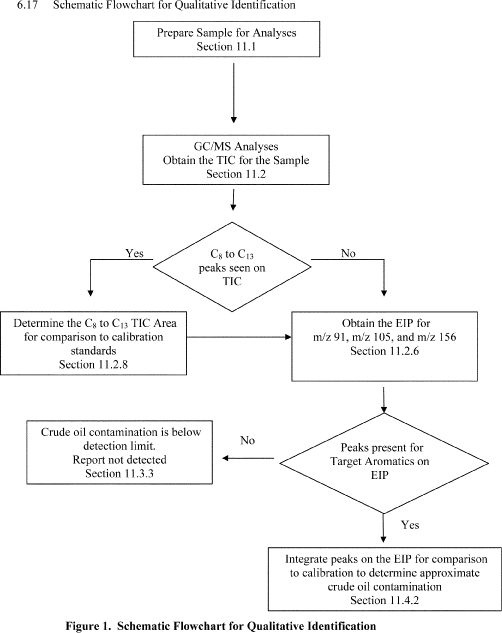 12.0
Calculations
12.0
Calculations
The concentration of oil in NAFs drilling fluids shall be computed relative to peak areas between C8 and C13 (using the Total Area Integration method) or total peak areas from extracted ion profiles (using the Extracted Ion Profile Method). In either case, there is a measurable amount of peak area, even in clean drilling fluid samples, due to spurious peaks and electrometer “noise” that contributes to the total signal measured using either of the quantification methods. In this procedure, a correction for this signal is applied, using the blank or clean sample correction technique described in American Society for Testing Materials (ASTM) Method D-3328-90, Comparison of Waterborne Oil by Gas Chromatography. In this method, the “oil equivalents” measured in a blank sample by total area gas chromatography are subtracted from that determined for a field sample to arrive at the most accurate measure of oil residue in the authentic sample.
12.1 Total Area Integration Method
12.1.1 Using C8 to C13 TIC area, the TCB area in the clean NAF sample and the TIC linear regression curve, compute the oil equivalent concentration of the C8 to C13 retention time range in the clean NAF.
Note:The actual TIC area of the C8 to C13 is equal to the C8 to C13 area minus the area of the TCB.
12.1.2 Using the corresponding information for the authentic sample, compute the oil equivalent concentration of the C8 to C13 retention time range in the authentic sample.
12.1.3 Calculate the concentration (% oil) of oil in the sample by subtracting the oil equivalent concentration (% oil) found in the clean NAF from the oil equivalent concentration (% oil) found in the authentic sample.
12.2 EIP Area Integration Method
12.2.1 Using either m/z 91 or 105 EIP areas, the TCB area in the clean NAF sample, and the appropriate EIP linear regression curve, compute the oil equivalent concentration of the in the clean NAF.
12.2.2 Using the corresponding information for the authentic sample, compute its oil equivalent concentration.
12.2.3 Calculate the concentration (% oil) of oil in the sample by subtracting the oil equivalent concentration (% oil) found in the clean NAF from the oil equivalent concentration (% oil) found in the authentic sample.
13.0 Method Performance13.1 Specification in this method are adopted from EPA Method 1663, Differentiation of Diesel and Crude Oil by GC/FID (Reference 16.5).
13.2 Single laboratory method performance using an Internal Olefin (IO) drilling fluid fortified at 0.5% oil using a 35 API gravity oil was:
Precision and accuracy 94 ±4% Accuracy interval - 86.3% to 102% Relative percent difference in duplicate analysis - 6.2% 14.0 Pollution Prevention14.1 The solvent used in this method poses little threat to the environment when recycled and managed properly.
15.0 Waste Management15.1 It is the laboratory's responsibility to comply with all federal, state, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restriction, and to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance with all sewage discharge permits and regulations is also required.
15.2 All authentic samples (drilling fluids) failing the RPE (fluorescence) test (indicated by the presence of fluorescence) shall be retained and classified as contaminated samples. Treatment and ultimate fate of these samples is not outlined in this SOP.
15.3 For further information on waste management, consult “The Waste Management Manual for Laboratory Personnel”, and “Less is Better: Laboratory Chemical Management for Waste Reduction”, both available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street NW, Washington, DC 20036.
16.0 References16.1 Carcinogens - “Working With Carcinogens.” Department of Health, Education, and Welfare, Public Health Service, Centers for Disease Control (available through National Technical Information Systems, 5285 Port Royal Road, Springfield, VA 22161, document no. PB-277256): August 1977.
16.2 “OSHA Safety and Health Standards, General Industry [29 CFR 1910], Revised.” Occupational Safety and Health Administration, OSHA 2206. Washington, DC: January 1976.
16.3 “Handbook of Analytical Quality Control in Water and Wastewater Laboratories.” USEPA, EMSSL-CI, EPA-600/4-79-019. Cincinnati, OH: March 1979.
16.4 “Method 1663, Differentiation of Diesel and Crude Oil by GC/FID, Methods for the Determination of Diesel, Mineral, and Crude Oils in Offshore Oil and Gas Industry Discharges, EPA 821-R-92-008, Office of Water Engineering and Analysis Division, Washington, DC: December 1992.
[66 FR 6901, Jan. 22, 2001, as amended at 77 FR 29843, May 18, 2012]Appendix 6 to Subpart A of Part 435 - Reverse Phase Extraction (RPE) Method for Detection of Oil Contamination in Non-Aqueous Drilling Fluids (NAF) (GC/MS) (EPA Method 1670)
40:32.0.1.1.11.1.4.7.12 :
Appendix 6 to Subpart A of Part 435 - Reverse Phase Extraction (RPE) Method for Detection of Oil Contamination in Non-Aqueous Drilling Fluids (NAF) (GC/MS) (EPA Method 1670) 1.0 Scope and Application1.1 This method is used for determination of crude or formation oil, or other petroleum oil contamination, in non-aqueous drilling fluids (NAFs).
1.2 This method is intended as a positive/negative test to determine a presence of crude oil in NAF prior to discharging drill cuttings from offshore production platforms.
1.3 This method is for use in the Environmental Protection Agency's (EPA's) survey and monitoring programs under the Clean Water Act, including monitoring of compliance with the Gulf of Mexico NPDES General Permit for monitoring of oil contamination in drilling fluids.
1.4 This method has been designed to show positive contamination for 5% of representative crude oils at a concentration of 0.1% in drilling fluid (vol/vol), 50% of representative crude oils at a concentration of 0.5%, and 95% of representative crude oils at a concentration of 1%.
1.5 Any modification of this method, beyond those expressly permitted, shall be considered a major modification subject to application and approval of alternate test procedures under 40 CFR parts 136.4 and 136.5.
1.6 Each laboratory that uses this method must demonstrate the ability to generate acceptable results using the procedure in Section 9.2 of this appendix.
2.0 Summary of Method2.1 An aliquot of drilling fluid is extracted using isopropyl alcohol.
2.2 The mixture is allowed to settle and then filtered to separate out residual solids.
2.3 An aliquot of the filtered extract is charged onto a reverse phase extraction (RPE) cartridge.
2.4 The cartridge is eluted with isopropyl alcohol.
2.5 Crude oil contaminates are retained on the cartridge and their presence (or absence) is detected based on observed fluorescence using a black light.
3.0 Definitions3.1 A NAF is one in which the continuous phase is a water immiscible fluid such as an oleaginous material (e.g., mineral oil, enhance mineral oil, paraffinic oil, or synthetic material such as olefins and vegetable esters).
4.0 Interferences4.1 Solvents, reagents, glassware, and other sample-processing hardware may yield artifacts that affect results. Specific selection of reagents and purification of solvents may be required.
4.2 All materials used in the analysis shall be demonstrated to be free from interferences under the conditions of analysis by running laboratory reagent blanks as described in Section 9.5 of this appendix.
5.0 Safety5.1 The toxicity or carcinogenicity of each reagent used in this method has not been precisely determined; however, each chemical shall be treated as a potential health hazard. Exposure to these chemicals should be reduced to the lowest possible level. Material Safety Data Sheets (MSDSs) shall be available for all reagents.
5.2 Isopropyl alcohol is flammable and should be used in a well-ventilated area.
5.3 Unknown samples may contain high concentration of volatile toxic compounds. Sample containers should be opened in a hood and handled with gloves to prevent exposure. In addition, all sample preparation should be conducted in a well-ventilated area to limit the potential exposure to harmful contaminants. Drilling fluid samples should be handled with the same precautions used in the drilling fluid handling areas of the drilling rig.
5.4 This method does not address all safety issues associated with its use. The laboratory is responsible for maintaining a safe work environment and a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material safety data sheets (MSDSs) shall be available to all personnel involved in these analyses. Additional information on laboratory safety can be found in References 16.1-16.2.
6.0 Equipment and Supplies Note:Brand names, suppliers, and part numbers are for illustrative purposes only. No endorsement is implied. Equivalent performance may be achieved using apparatus and materials other than those specified here, but demonstration of equivalent performance that meets the requirements of this method is the responsibility of the laboratory.
6.1 Sampling equipment.
6.1.1 Sample collection bottles/jars - New, pre-cleaned bottles/jars, lot-certified to be free of artifacts. Glass preferable, plastic acceptable, wide mouth approximately 1-L, with Teflon-lined screw cap.
6.2 Equipment for glassware cleaning.
6.2.1 Laboratory sink.
6.2.2 Oven - Capable of maintaining a temperature within ±5 °C in the range of 100-250 °C.
6.3 Equipment for sample extraction.
6.3.1 Vials - Glass, 25 mL and 4 mL, with Teflon-lined screw caps, baked at 200-250 °C for 1-h minimum prior to use.
6.3.2 Gas-tight syringes - Glass, various sizes, 0.5 mL to 2.5 mL (if spiking of drilling fluids with oils is to occur).
6.3.3 Auto pipetters - various sizes, 0.1 mL, 0.5 mL, 1 to 5 mL delivery, and 10 mL delivery, with appropriate size disposable pipette tips, calibrated to within ±0.5%.
6.3.4 Glass stirring rod.
6.3.5 Vortex mixer.
6.3.6 Disposable syringes - Plastic, 5 mL.
6.3.7 Teflon syringe filter, 25-mm, 0.45 µm pore size - Acrodisc ® CR Teflon (or equivalent).
6.3.8 Reverse Phase Extraction C18 Cartridge - Waters Sep-Pak ®Plus, C18 Cartridge, 360 mg of sorbent (or equivalent).
6.3.9 SPE vacuum manifold - Supelco Brand, 12 unit (or equivalent). Used as support for cartridge/syringe assembly only. Vacuum apparatus not required.
6.4 Equipment for fluorescence detection.
6.4.1 Black light - UV Lamp, Model UVG 11, Mineral Light Lamp, Shortwave 254 nm, or Longwave 365 nm, 15 volts, 60 Hz, 0.16 amps (or equivalent).
6.4.2 Black box - cartridge viewing area. A commercially available ultraviolet viewing cabinet with viewing lamp, or alternatively, a cardboard box or equivalent, approximately 14″ × 7.5″ × 7.5″ in size and painted flat black inside. Lamp positioned in fitted and sealed slot in center on top of box. Sample cartridges sit in a tray, ca. 6″ from lamp. Cardboard flaps cut on top panel and side of front panel for sample viewing and sample cartridge introduction, respectively.
6.4.3 Viewing platform for cartridges. Simple support (hand made vial tray - black in color) for cartridges so that they do not move during the fluorescence testing.
7.0 Reagents and Standards7.1 Isopropyl alcohol - 99% purity.
7.2 NAF - Appropriate NAF as sent from the supplier (has not been circulated downhole). Use the clean NAF corresponding to the NAF being used in the current drilling operation.
7.3 Standard crude oil - NIST SRM 1582 petroleum crude oil.
8.0 Sample Collection, Preservation, and Storage8.1 Collect approximately one liter of representative sample (NAF, which has been circulated downhole) in a glass bottle or jar. Cover with a Teflon lined cap. To allow for a potential need to re-analyze and/or re-process the sample, it is recommended that a second sample aliquot be collected.
8.2 Label the sample appropriately.
8.3 All samples must be refrigerated at 0-4 °C from the time of collection until extraction (40 CFR part 136, Table II).
8.4 All samples must be analyzed within 28 days of the date and time of collection (40 CFR part 136, Table II).
9.0 Quality Control9.1 Each laboratory that uses this method is required to operate a formal quality assurance program (Reference 16.3). The minimum requirements of this program consist of an initial demonstration of laboratory capability, and ongoing analyses of blanks and spiked duplicates to assess accuracy and precision and to demonstrate continued performance. Each field sample is analyzed in duplicate to demonstrate representativeness.
9.1.1 The analyst shall make an initial demonstration of the ability to generate acceptable accuracy and precision with this method. This ability is established as described in Section 9.2 of this appendix.
9.1.2 Preparation and analysis of a set of spiked duplicate samples to document accuracy and precision. The procedure for the preparation and analysis of these samples is described in Section 9.4 of this appendix.
9.1.3 Analyses of laboratory reagent blanks are required to demonstrate freedom from contamination. The procedure and criteria for preparation and analysis of a reagent blank are described in Section 9.5 of this appendix.
9.1.4 The laboratory shall maintain records to define the quality of the data that is generated.
9.1.5 Accompanying QC for the determination of oil in NAF is required per analytical batch. An analytical batch is a set of samples extracted at the same time, to a maximum of 10 samples. Each analytical batch of 10 or fewer samples must be accompanied by a laboratory reagent blank (Section 9.5 of this appendix), corresponding NAF reference blanks (Section 9.6 of this appendix), a set of spiked duplicate samples blank (Section 9.4 of this appendix), and duplicate analysis of each field sample. If greater than 10 samples are to be extracted at one time, the samples must be separated into analytical batches of 10 or fewer samples.
9.2 Initial demonstration of laboratory capability. To demonstrate the capability to perform the test, the analyst shall analyze two representative unused drilling fluids (e.g., internal olefin-based drilling fluid, vegetable ester-based drilling fluid), each prepared separately containing 0.1%, 1%, and 2% or a representative oil. Each drilling fluid/concentration combination shall be analyzed 10 times, and successful demonstration will yield the following average results for the data set:
0.1% oil - Detected in <20% of samples 1% oil - Detected in >75% of samples 2% oil - Detected in >90% of samples9.3 Sample duplicates.
9.3.1 The laboratory shall prepare and analyze (Section 11.2 and 11.4 of this appendix) each authentic sample in duplicate, from a given sampling site or, if for compliance monitoring, from a given discharge.
9.3.2 The duplicate samples must be compared versus the prepared corresponding NAF blank.
9.3.3 Prepare and analyze the duplicate samples according to procedures outlined in Section 11 of this appendix.
9.3.4 The results of the duplicate analyses are acceptable if each of the results give the same response (fluorescence or no fluorescence). If the results are different, sample non-homogenicity issues may be a concern. Prepare the samples again, ensuring a well-mixed sample prior to extraction. Analyze the samples once again.
9.3.5 If different results are obtained for the duplicate a second time, the analytical system is judged to be out of control and the problem shall be identified and corrected, and the samples re-analyzed.
9.4 Spiked duplicates - Laboratory prepared spiked duplicates are analyzed to demonstrate acceptable accuracy and precision.
9.4.1 Preparation and analysis of a set of spiked duplicate samples with each set of no more than 10 field samples is required to demonstrate method accuracy and precision and to monitor matrix interferences (interferences caused by the sample matrix). A field NAF sample expected to contain less than 0.5% crude oil (and documented to not fluoresce as part of the sample batch analysis) shall be spiked with 1% (by volume) of suitable reference crude oil and analyzed as field samples, as described in Section 11 of this appendix. If no low-level drilling fluid is available, then the unused NAF can be used as the drilling fluid sample.
9.5 Laboratory reagent blanks - Laboratory reagent blanks are analyzed to demonstrate freedom from contamination.
9.5.1 A reagent blank is prepared by passing 4 mL of the isopropyl alcohol through a Teflon syringe filter and collecting the filtrate in a 4-mL glass vial. A Sep Pak ® C18 cartridge is then preconditioned with 3 mL of isopropyl alcohol. A 0.5-mL aliquot of the filtered isopropyl alcohol is added to the syringe barrel along with 3.0 mL of isopropyl alcohol. The solvent is passed through the preconditioned Sep Pak ® cartridge. An additional 2-mL of isopropyl alcohol is eluted through the cartridge. The cartridge is now considered the “reagent blank” cartridge and is ready for viewing (analysis). Check the reagent blank cartridge under the black light for fluorescence. If the isopropyl alcohol and filter are clean, no fluorescence will be observed.
9.5.2 If fluorescence is detected in the reagent blank cartridge, analysis of the samples is halted until the source of contamination is eliminated and a prepared reagent blank shows no fluorescence under a black light. All samples shall be associated with an uncontaminated method blank before the results may be reported for regulatory compliance purposes.
9.6 NAF reference blanks - NAF reference blanks are prepared from the NAFs sent from the supplier (NAF that has not been circulated downhole) and used as the reference when viewing the fluorescence of the test samples.
9.6.1 A NAF reference blank is prepared identically to the authentic samples. Place a 0.1 mL aliquot of the “clean” NAF into a 25-mL glass vial. Add 10 mL of isopropyl alcohol to the vial. Cap the vial. Vortex the vial for approximately 10 sec. Allow the solids to settle for approximately 15 minutes. Using a 5-mL syringe, draw up 4 mL of the extract and filter it through a PTFE syringe filter, collecting the filtrate in a 4-mL glass vial. Precondition a Sep Pak ® C18 cartridge with 3 mL of isopropyl alcohol. Add a 0.5-mL aliquot of the filtered extract to the syringe barrel along with 3.0 mL of isopropyl alcohol. Pass the extract and solvent through the preconditioned Sep Pak ® cartridge. Pass an additional 2-mL of isopropyl alcohol through the cartridge. The cartridge is now considered the NAF blank cartridge and is ready for viewing (analysis). This cartridge is used as the reference cartridge for determining the absence or presence of fluorescence in all authentic drilling fluid samples that originate from the same NAF. That is, the specific NAF reference blank cartridge is put under the black light along with a prepared cartridge of an authentic sample originating from the same NAF material. The fluorescence or absence of fluorescence in the authentic sample cartridge is determined relative to the NAF reference cartridge.
9.6.2 Positive control solution, equivalent to 1% crude oil contaminated mud extract, is prepared by dissolving 87 mg of standard crude oil into 10.00 mL of methylene chloride. Then mix 40 µL of this solution into 10.00 mL of IPA. Transfer 0.5 mL of this solution into a preconditioned C18 cartridge, followed by 2 ml of IPA.
10.0 Calibration and Standardization10.1 Calibration and standardization methods are not employed for this procedure.
11.0 ProcedureThis method is a screening-level test. Precise and accurate results can be obtained only by strict adherence to all details.
11.1 Preparation of the analytical batch.
11.1.1 Bring the analytical batch of samples to room temperature.
11.1.2 Using a large glass stirring rod, mix the authentic sample thoroughly.
11.1.3 Using a large glass stirring rod, mix the clean NAF (sent from the supplier) thoroughly.
11.2 Extraction.
11.2.1 Using an automatic positive displacement pipetter and a disposable pipette tip transfer 0.1-mL of the authentic sample into a 25-mL vial.
11.2.2 Using an automatic pipetter and a disposable pipette tip dispense a 10-mL aliquot of solvent grade isopropyl alcohol (IPA) into the 25 mL vial.
11.2.3 Cap the vial and vortex the vial for ca. 10-15 seconds.
11.2.4 Let the sample extract stand for approximately 5 minutes, allowing the solids to separate.
11.2.5 Using a 5-mL disposable plastic syringe remove 4 mL of the extract from the 25-mL vial.
11.2.6 Filter 4 mL of extract through a Teflon syringe filter (25-mm diameter, 0.45 µm pore size), collecting the filtrate in a labeled 4-mL vial.
11.2.7 Dispose of the PFTE syringe filter.
11.2.8 Using a black permanent marker, label a Sep Pak ® C18 cartridge with the sample identification.
11.2.9 Place the labeled Sep Pak ® C18 cartridge onto the head of a SPE vacuum manifold.
11.2.10 Using a 5-mL disposable plastic syringe, draw up exactly 3-mL (air free) of isopropyl alcohol.
11.2.11 Attach the syringe tip to the top of the C18 cartridge.
11.2.12 Condition the C18 cartridge with the 3-mL of isopropyl alcohol by depressing the plunger slowly.
Note:Depress the plunger just to the point when no liquid remains in the syringe barrel. Do not force air through the cartridge. Collect the eluate in a waste vial.
11.2.13 Remove the syringe temporarily from the top of the cartridge, then remove the plunger, and finally reattach the syringe barrel to the top of the C18 cartridge.
11.2.14 Using automatic pipetters and disposable pipette tips, transfer 0.5 mL of the filtered extract into the syringe barrel, followed by a 3.0-mL transfer of isopropyl alcohol to the syringe barrel.
11.2.15 Insert the plunger and slowly depress it to pass only the extract and solvent through the preconditioned C18 cartridge.
Note:Depress the plunger just to the point when no liquid remains in the syringe barrel. Do not force air through the cartridge. Collect the eluate in a waste vial.
11.2.16 Remove the syringe temporarily from the top of the cartridge, then remove the plunger, and finally reattach the syringe barrel to the top of the C18 cartridge.
11.2.17 Using an automatic pipetter and disposable pipette tip, transfer 2.0 mL of isopropyl alcohol to the syringe barrel.
11.2.18 Insert the plunger and slowly depress it to pass the solvent through the C18 cartridge.
Note:Depress the plunger just to the point when no liquid remains in the syringe barrel. Do not force air through the cartridge. Collect the eluate in a waste vial.
11.2.19 Remove the syringe and labeled C18 cartridge from the top of the SPE vacuum manifold.
11.2.20 Prepare a reagent blank according to the procedures outlined in Section 9.5 of this appendix.
11.2.21 Prepare the necessary NAF reference blanks for each type of NAF encountered in the field samples according to the procedures outlined in Section 9.6 of this appendix.
11.2.22 Prepare the positive control (1% crude oil equivalent) according to Section 9.6.2 of this appendix.
11.3 Reagent blank fluorescence testing.
11.3.1 Place the reagent blank cartridge in a black box, under a black light.
11.3.2 Determine the presence or absence of fluorescence for the reagent blank cartridge. If fluorescence is detected in the blank, analysis of the samples is halted until the source of contamination is eliminated and a prepared reagent blank shows no fluorescence under a black light. All samples must be associated with an uncontaminated method blank before the results may be reported for regulatory compliance purposes.
11.4 Sample fluorescence testing.
11.4.1 Place the respective NAF reference blank (Section 9.6 of this appendix) onto the tray inside the black box.
11.4.2 Place the authentic field sample cartridge (derived from the same NAF as the NAF reference blank) onto the tray, adjacent and to the right of the NAF reference blank.
11.4.3 Turn on the black light.
11.4.4 Compare the fluorescence of the sample cartridge with that of the negative control cartridge (NAF blank, Section 9.6.1 of this appendix) and positive control cartridge (1% crude oil equivalent, Section 9.6.2 of this appendix).
11.4.5 If the fluorescence of the sample cartridge is equal to or brighter than the positive control cartridge (1% crude oil equivalent, Section 9.6.2 of this appendix), the sample is considered contaminated. Otherwise, the sample is clean.
12.0 Data Analysis and CalculationsSpecific data analysis techniques and calculations are not performed in this SOP.
13.0 Method PerformanceThis method was validated through a single laboratory study, conducted with rigorous statistical experimental design and interpretation (Reference 16.4).
14.0 Pollution Prevention14.1 The solvent used in this method poses little threat to the environment when recycled and managed properly.
15.0 Waste Management15.1 It is the laboratory's responsibility to comply with all Federal, State, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restriction, and to protect the air, water, and land by minimizing and controlling all releases from bench operations. Compliance with all sewage discharge permits and regulations is also required.
15.2 All authentic samples (drilling fluids) failing the fluorescence test (indicated by the presence of fluorescence) shall be retained and classified as contaminated samples. Treatment and ultimate fate of these samples is not outlined in this SOP.
15.3 For further information on waste management, consult “The Waste Management Manual for Laboratory Personnel,” and “Less is Better: Laboratory Chemical Management for Waste Reduction,” both available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street, NW, Washington, DC 20036.
16.0 References16.1 “Carcinogen - Working with Carcinogens,” Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, August 1977.
16.2 “OSHA Safety and Health Standards, General Industry,” (29 CFR 1910), Occupational Safety and Health Administration, OSHA 2206 (Revised, January 1976).
16.3 “Handbook of Analytical Quality Control in Water and Wastewater Laboratories,” USEPA, EMSL-Ci, Cincinnati, OH 45268, EPA-600/4-79-019, March 1979.
16.4 Report of the Laboratory Evaluation of Static Sheen Test Replacements - Reverse Phase Extraction (RPE) Method for Detecting Oil Contamination in Synthetic Based Mud (SBM). October 1998. Available from API, 1220 L Street, NW, Washington, DC 20005-4070, 202-682-8000.
[66 FR 6901, Jan. 22, 2001; 66 FR 30811, June 8, 2001]Appendix 7 to Subpart A of Part 435 - Determination of the Amount of Non-Aqueous Drilling Fluid (NAF) Base Fluid From Drill Cuttings by a Retort Chamber (Derived From API Recommended Practice 13B-2) (EPA Method 1674)
40:32.0.1.1.11.1.4.7.13 :
Appendix 7 to Subpart A of Part 435 - Determination of the Amount of Non-Aqueous Drilling Fluid (NAF) Base Fluid From Drill Cuttings by a Retort Chamber (Derived From API Recommended Practice 13B-2) (EPA Method 1674) 1. Descriptiona. This procedure is specifically intended to measure the amount of non-aqueous drilling fluid (NAF) base fluid from cuttings generated during a drilling operation. This procedure is a retort test which measures all oily material (NAF base fluid) and water released from a cuttings sample when heated in a calibrated and properly operating “Retort” instrument.
b. In this retort test a known mass of cuttings is heated in the retort chamber to vaporize the liquids associated with the sample. The NAF base fluid and water vapors are then condensed, collected, and measured in a precision graduated receiver.
Note:Obtaining a representative sample requires special attention to the details of sample handling (e.g., location, method, frequency). See Addendum A and B for minimum requirements for collecting representative samples. Additional sampling procedures in a given area may be specified by the NPDES permit controlling authority.
2. Equipmenta. Retort instrument - The recommended retort instrument has a 50-cm 3 volume with an external heating jacket.
Retort Specifications:
1. Retort assembly - retort body, cup and lid.
(a) Material: 303 stainless steel or equivalent.
(b) Volume: Retort cup with lid.
Cup Volume: 50-cm 3.
Precision: ±0.25-cm 3.
2. Condenser - capable of cooling the oil and water vapors below their liquification temperature.
3. Heating jacket - nominal 350 watts.
4. Temperature control - capable of limiting temperature of retort to at least 930 °F (500 °C) and enough to boil off all NAFs.
b. Liquid receiver (10-cm 3, 20-cm 3) - the 10-cm 3 and 20-cm 3 receivers are specially designed cylindrical glassware with rounded bottom to facilitate cleaning and funnel-shaped top to catch falling drops. For compliance monitoring under the NPDES program, the analyst shall use the 10-cm 3 liquid receiver with 0.1 ml graduations to achieve greater accuracy.
1. Receiver specifications:
Total volume: 10-cm 3, 20-cm 3.
Precision (0 to 100%): ±0.05 cm 3, ±0.05 cm 3.
Outside diameter: 10-mm, 13-mm.
Wall thickness: 1.5 ±0.1mm, 1.2 ±0.1mm.
Frequency of graduation marks (0 to 100%): 0.10-cm 3, 0.10-cm 3.
Calibration: To contain “TC” @ 20 °C.
Scale: cm 3, cm 3
2. Material - Pyrex ® or equivalent glass.
c. Toploading balance - capable of weighing 2000 g and precision of at least 0.1 g. Unless motion is a problem, the analyst shall use an electronic balance. Where motion is a problem, the analyst may use a triple beam balance.
d. Fine steel wool (No. 000) - for packing retort body.
e. Thread sealant lubricant: high temperature lubricant, e.g. Never-Seez ® or equivalent.
f. Pipe cleaners - to clean condenser and retort stem.
g. Brush - to clean receivers.
h. Retort spatula - to clean retort cup.
i. Corkscrew - to remove spent steel wool.
3. Procedurea. Clean and dry the retort assembly and condenser.
b. Pack the retort body with steel wool.
c. Apply lubricant/sealant to threads of retort cup and retort stem.
d. Weigh and record the total mass of the retort cup, lid, and retort body with steel wool. This is mass (A), grams.
e. Collect a representative cuttings sample (see note in section 1 of this appendix).
f. Partially fill the retort cup with cuttings and place the lid on the cup.
g. Screw the retort cup (with lid) onto the retort body, weigh and record the total mass. This is mass (B), grams.
h. Attach the condenser. Place the retort assembly into the heating jacket.
i. Weigh and record the mass of the clean and dry liquid receiver. This is mass (C), grams. Place the receiver below condenser outlet.
j. Turn on the retort. Allow it to run a minimum of 1 hour.
Note:If solids boil over into receiver, the test shall be rerun. Pack the retort body with a greater amount of steel wool and repeat the test.
k. Remove the liquid receiver. Allow it to cool. Record the volume of water recovered. This is (V), cm 3.
Note:If an emulsion interface is present between the oil and water phases, heating the interface may break the emulsion. As a suggestion, remove the retort assembly from the heating jacket by grasping the condenser. Carefully heat the receiver along the emulsion band by gently touching the receiver for short intervals with the hot retort assembly. Avoid boiling the liquids. After the emulsion interface is broken, allow the liquid receiver to cool. Read the water volume at the lowest point of the meniscus.
l. Weigh and record the mass of the receiver and its liquid contents (oil plus water). This is mass (D), grams.
m. Turn off the retort. Remove the retort assembly and condenser from the heating jacket and allow them to cool. Remove the condenser.
n. Weigh and record the mass of the cooled retort assembly without the condenser. This is mass (E), grams.
o. Clean the retort assembly and condenser.
4. Calculationsa. Calculate the mass of oil (NAF base fluid) from the cuttings as follows:
1. Mass of the wet cuttings sample (Mw) equals the mass of the retort assembly with the wet cuttings sample (B) minus the mass of the empty retort assembly (A).
Mw = B−A [1]2. Mass of the dry retorted cuttings (MD) equals the mass of the cooled retort assembly (E) minus the mass of the empty retort assembly (A).
MD = E−A [2]3. Mass of the NAF base fluid (MBF) equals the mass of the liquid receiver with its contents (D) minus the sum of the mass of the dry receiver (C) and the mass of the water (V).
MBF = D−(C + V) [3] Note:Assuming the density of water is 1 g/cm 3, the volume of water is equivalent to the mass of the water.
b. Mass balance requirement:
The sum of MD, MBF, and V shall be within 5% of the mass of the wet sample.
(MD + MBF + V)/Mw = 0.95 to 1.05 [4]The procedure shall be repeated if this requirement is not met.
c. Reporting oil from cuttings:
1. Assume that all oil recovered is NAF base fluid.
2. The mass percent NAF base fluid retained on the cuttings (%BFi) for the sampled discharge “i” is equal to 100 times the mass of the NAF base fluid (MBF) divided by the mass of the wet cuttings sample (Mw).
%BFi = (MBF/Mw) × 100 [5]Operators discharging small volume NAF-cuttings discharges which do not occur during a NAF-cuttings discharge sampling interval (i.e., displaced interfaces, accumulated solids in sand traps, pit clean-out solids, or centrifuge discharges while cutting mud weight) shall either: (a) Measure the mass percent NAF base fluid retained on the cuttings (%BFSVD) for each small volume NAF-cuttings discharges; or (b) use a default value of 25% NAF base fluid retained on the cuttings.
3. The mass percent NAF base fluid retained on the cuttings is determined for all cuttings wastestreams and includes fines discharges and any accumulated solids discharged [see Section 4.c.6 of this appendix for procedures on measuring or estimating the mass percent NAF base fluid retained on the cuttings (%BF) for dual gradient drilling seafloor discharges performed to ensure proper operation of subsea pumps].
4. A mass NAF-cuttings discharge fraction (X, unitless) is calculated for all NAF-cuttings, fines, or accumulated solids discharges every time a set of retorts is performed (see Section 4.c.6 of this appendix for procedures on measuring or estimating the mass NAF-cuttings discharge fraction (X) for dual gradient drilling seafloor discharges performed to ensure proper operation of subsea pumps). The mass NAF-cuttings discharge fraction (X) combines the mass of NAF-cuttings, fines, or accumulated solids discharged from a particular discharge over a set period of time with the total mass of NAF-cuttings, fines, or accumulated solids discharged into the ocean during the same period of time (see Addendum A and B of this appendix). The mass NAF-cuttings discharge fraction (X) for each discharge is calculated by direct measurement as:
Xi = (Fi)/(G) [6] where: Xi = Mass NAF-cuttings discharge fraction for NAF-cuttings, fines, or accumulated solids discharge “i”, (unitless) Fi = Mass of NAF-cuttings discharged from NAF-cuttings, fines, or accumulated solids discharge “i” over a specified period of time (see Addendum A and B of this appendix), (kg) G = Mass of all NAF-cuttings discharges into the ocean during the same period of time as used to calculate Fi, (kg)If an operator has more than one point of NAF-cuttings discharge, the mass faction (Xi) must be determined by: (a) Direct measurement (see Equation 6 of this appendix); (b) using the following default values of 0.85 and 0.15 for the cuttings dryer (e.g., horizontal centrifuge, vertical centrifuge, squeeze press, High-G linear shakers) and fines removal unit (e.g., decanting centrifuges, mud cleaners), respectively, when the operator is only discharging from the cuttings dryer and the fines removal unit; or (c) using direct measurement of “Fi” (see Equation 6 of this appendix) for fines and accumulated solids, using Equation 6A of this appendix to calculate “GEST” for use as “G” in Equation 6 of this Appendix, and calculating the mass (kg) of NAF-cuttings discharged from the cuttings dryer (Fi) as the difference between the mass of “GEST” calculated in Equation 6A of this appendix (kg) and the sum of all fines and accumulated solids mass directly measured (kg) (see Equation 6 of this appendix).
GEST = Estimated mass of all NAF-cuttings discharges into the ocean during the same period of time as used to calculate Fi (see Equation 6 of this appendix), (kg) [6A] where: GEST = Hole Volume (bbl) × (396.9 kg/bbl) × (1 + Z/100) Z = The base fluid retained on cuttings limitation or standard (%) which apply to the NAF being discharge (see §§ 435.13. and 435.15). Hole Volume (bbl) = [Cross-Section Area of NAF interval (in 2)] × Average Rate of Penetration (feet/hr) × period of time (min) used to calculate Fi (see Equation 6 of this appendix) × (1 hr/60 min) × (1 bbl/5.61 ft 3) × (1 ft/12 in) 2 Cross-Section Area of NAF interval (in 2) = (3.14 × [Bit Diameter (in)] 2)/4 Bit Diameter (in) = Diameter of drilling bit for the NAF interval producing drilling cuttings during the same period of time as used to calculate Fi (see Equation 6 of this appendix) Average Rate of Penetration (feet/hr) = Arithmetic average of rate of penetration into the formation during the same period of time as used to calculate Fi (see Equation 6 of this appendix) Note:Operators with one NAF-cuttings discharge may set the mass NAF-cuttings discharge fraction (Xi) equal to 1.0.
5. Each NAF-cuttings, fines, or accumulated solids discharge has an associated mass percent NAF base fluid retained on cuttings value (%BF) and mass NAF-cuttings discharge fraction (X) each time a set of retorts is performed. A single total mass percent NAF base fluid retained on cuttings value (%BFT) is calculated every time a set of retorts is performed. The single total mass percent NAF base fluid retained on cuttings value (%BFT) is calculated as:
%BFT,j = Σ(Xi) × (%BFi) [7] where: %BFT,j = Total mass percent NAF base fluid retained on cuttings value for retort set “j” (unitless as percentage, %) Xi = Mass NAF-cuttings discharge fraction for NAF-cuttings, fines, or accumulated solids discharge “i”, (unitless) %BFi = Mass percent NAF base fluid retained on the cuttings for NAF-cuttings, fines, or accumulated solids discharge “i” , (unitless as percentage, %) Note:ΣXi = 1.
Operators with one NAF-cuttings discharge may set %BFT,j equal to %BFi.
6. Operators performing dual gradient drilling operations may require seafloor discharges of large cuttings (> 1/4′) to ensure the proper operation of subsea pumps (e.g., electrical submersible pumps). Operators performing dual gradient drilling operations which lead to seafloor discharges of large cuttings for the proper operation of subsea pumps shall either: (a) Measure the mass percent NAF base fluid retained on cuttings value (%BF) and mass NAF-cuttings discharge fraction (X) for seafloor discharges each time a set of retorts is performed; (b) use the following set of default values, (%BF = 14%; X = 0.15); or (c) use a combination of (a) and (b) (e.g., use a default value for %BF and measure X).
Additionally, operators performing dual gradient drilling operations which lead to seafloor discharges of large cuttings for the proper operation of subsea pumps shall also perform the following tasks:
(a) Use side scan sonar or shallow seismic to determine the presence of high density chemosynthetic communities. Chemosynthetic communities are assemblages of tube worms, clams, mussels, and bacterial mats that occur at natural hydrocarbon seeps or vents, generally in water depths of 500 meters or deeper. Seafloor discharges of large cuttings for the proper operation of subsea pumps shall not be permitted within 1000 feet of a high density chemosynthetic community.
(b) Seafloor discharges of large cuttings for the proper operation of subsea pumps shall be visually monitored and documented by a Remotely Operated Vehicle (ROV) within the tether limit (approximately 300 feet). The visual monitoring shall be conducted prior to each time the discharge point is relocated (cuttings discharge hose) and conducted along the same direction as the discharge hose position. Near-seabed currents shall be obtained at the time of the visual monitoring.
(c) Seafloor discharges of large cuttings for the proper operation of subsea pumps shall be directed within a 150 foot radius of the wellbore.
7. The weighted mass ratio averaged over all NAF well sections (%BFwell) is the compliance value that is compared with the “maximum weighted mass ratio averaged over all NAF well sections” BAT discharge limitations (see the table in § 435.13 and footnote 5 of the table in § 435.43) or the “maximum weighted mass ratio averaged over all NAF well sections” NSPS discharge limitations (see the table in § 435.15 and footnote 5 of the table in § 435.45). The weighted mass ratio averaged over all NAF well sections (%BFwell) is calculated as the arithmetic average of all total mass percent NAF base fluid retained on cuttings values (%BFT) and is given by the following expression:
%BFwell = [j = 1 to j = n Σ (%BFT,j)]/n [8] where: %BFwell = Weighted mass ratio averaged over all NAF well sections (unitless as percentage, %) %BFT,j = Total mass percent NAF base fluid retained on cuttings value for retort set “j” (unitless as percentage, %) n = Total number of retort sets performed over all NAF well sections (unitless)Small volume NAF-cuttings discharges which do not occur during a NAF-cuttings discharge sampling interval (i.e., displaced interfaces, accumulated solids in sand traps, pit clean-out solids, or centrifuge discharges while cutting mud weight) shall be mass averaged with the arithmetic average of all total mass percent NAF base fluid retained on cuttings values (see Equation 8 of this appendix). An additional sampling interval shall be added to the calculation of the weighted mass ratio averaged over all NAF well sections (%BFwell). The mass fraction of the small volume NAF-cuttings discharges (XSVD) will be determined by dividing the mass of the small volume NAF-cuttings discharges (FSVD) by the total mass of NAF-cuttings discharges for the well drilling operation (GWELL + FSVD).
XSVD = FSVD / (GWELL + FSVD) [9] where: XSVD = mass fraction of the small volume NAF-cuttings discharges (unitless) FSVD = mass of the small volume NAF-cuttings discharges (kg) GWELL = mass of total NAF-cuttings from the well (kg)The mass of small volume NAF-cuttings discharges (FSVD) shall be determined by multiplying the density of the small volume NAF-cuttings discharges (ρsvd) times the volume of the small volume NAF-cuttings discharges (VSVD).
FSVD = ρsvd × VSVD [10] where: FSVD = mass of small volume NAF-cuttings discharges (kg) ρsvd = density of the small volume NAF-cuttings discharges (kg/bbl) VSVD = volume of the small volume NAF-cuttings discharges (bbl)The density of the small volume NAF-cuttings discharges shall be measured. The volume of small volume discharges (VSVD) shall be either: (a) Be measured or (b) use default values of 10 bbl of SBF for each interface loss and 75 bbl of SBM for pit cleanout per well.
The total mass of NAF-cuttings discharges for the well (GWELL) shall be either: (a) Measured; or (b) calculated by multiplying 1.0 plus the arithmetic average of all total mass percent NAF base fluid retained on cuttings values [see Equation 8 of this appendix] times the total hole volume (VWELL) for all NAF well sections times a default value for the density the formation of 2.5 g/cm 3 (396.9 kg/bbl).
where: GWELL = total mass of NAF-cuttings discharges for the well (kg) [j = 1 to j = n Σ(%BFT,j)]/n = see Equation 8 of this appendix (unitless as a percentage) VWELL = total hole volume (VWELL) for all NAF well sections (bbl)The total hole volume of NAF well sections (VWELL) will be calculated as:
For wells where small volume discharges associated with cuttings are made, %BFWELL becomes:
Note:See Addendum A and B to determine the sampling frequency to determine the total number of retort sets required for all NAF well sections.
8. The total number of retort sets (n) is increased by 1 for each sampling interval (see Section 2.4, Addendum A of this appendix) when all NAF cuttings, fines, or accumulated solids for that sampling interval are retained for no discharge. A zero discharge interval shall be at least 500 feet up to a maximum of three per day. This action has the effect of setting the total mass percent NAF base fluid retained on cuttings value (%BFT) at zero for that NAF sampling interval when all NAF cuttings, fines, or accumulated solids are retained for no discharge.
9. Operators that elect to use the Best Management Practices (BMPs) for NAF-cuttings shall use the procedures outlined in Addendum B.
Addendum A to Appendix 7 to Subpart A of Part 435 - Sampling of Cuttings Discharge Streams for use with API Recommended Practice 13B-2 1.0 Sampling Locations1.1 Each NAF-cuttings waste stream that discharges into the ocean shall be sampled and analyzed as detailed in appendix 7. NAF-cuttings discharges to the ocean may include discharges from primary shakers, secondary shakers, cuttings dryer, fines removal unit, accumulated solids, and any other cuttings separation device whose NAF-cuttings waste is discharged to the ocean. NAF-cuttings wastestreams not directly discharged to the ocean (e.g., NAF-cuttings generated from shake shakers and sent to a cuttings dryer for additional processing) do not require sampling and analysis.
1.2 The collected samples shall be representative of each NAF-cuttings discharge. Operators shall conduct sampling to avoid the serious consequences of error (i.e., bias or inaccuracy). Operators shall collect NAF-cuttings samples near the point of origin and before the solids and liquid fractions of the stream have a chance to separate from one another. For example, operators shall collect shale shaker NAF-cuttings samples at the point where NAF-cuttings are coming off the shale shaker and not from a holding container downstream where separation of larger particles from the liquid can take place.
1.3 Operators shall provide a simple schematic diagram of the solids control system and sample locations to the NPDES permit controlling authority.
2.0 Type of Sample and Sampling Frequency2.1 Each NAF-cuttings, fines, or accumulated solids discharge has an associated mass percent NAF base fluid retained on cuttings value (%BF) and mass NAF-cuttings discharge fraction (X) for each sampling interval (see Section 2.4 of this addendum). Operators shall collect a single discrete NAF-cuttings sample for each NAF-cuttings waste stream discharged to the ocean during every sampling interval.
2.2 Operators shall use measured depth in feet from the Kelly bushing when samples are collected.
2.3 The NAF-cuttings samples collected for the mass fraction analysis (see Equation 6, appendix 7 of subpart A of this part) shall also be used for the retort analysis (see Equations 1-5, appendix 7 of subpart A of this part).
2.4 Operators shall collect and analyze at least one set of NAF-cuttings samples per day while discharging. Operators engaged in fast drilling (i.e., greater than 500 linear NAF feet advancement of drill bit per day) shall collect and analyze one set of NAF-cuttings samples per 500 linear NAF feet of footage drilled. Operators are not required to collect and analyze more than three sets of NAF-cuttings samples per day (i.e., three sampling intervals). Operators performing zero discharge of all NAF-cuttings (i.e., all NAF cuttings, fines, or accumulated solids retained for no discharge) shall use the following periods to count sampling intervals: (1) One sampling interval per day when drilling is less than 500 linear NAF feet advancement of drill bit per day; and (2) one sampling interval per 500 linear NAF feet of footage drilled with a maximum of three sampling intervals per day.
2.5 The operator shall measure the individual masses (Fi, kg) and sum total mass (G, kg) (see Equation 6, appendix 7 of subpart A of this part) over a representative period of time (e.g., <10 minutes) during steady-state conditions for each sampling interval (see Section 2.4 of this addendum). The operator shall ensure that all NAF-cuttings are capture for mass analysis during the same sampling time period (e.g., <10 minutes) at approximately the same time (i.e., all individual mass samples collected within one hour of each other).
2.6 Operators using Best Management Practices (BMPs) to control NAF-cuttings discharges shall follow the procedures in Addendum B to appendix 7 of subpart A of 40 CFR 435.
3.0 Sample Size and Handling3.1 The volume of each sample depends on the volumetric flow rate (cm 3/s) of the NAF-cuttings stream and the sampling time period (e.g., <10 minutes). Consequently, different solids control equipment units producing different NAF-cuttings waste streams at different volumetric flow rates will produce different size samples for the same period of time. Operators shall use appropriately sized sample containers for each NAF-cuttings waste stream to ensure no NAF-cuttings are spilled during sample collection. Operators shall use the same time period (e.g., <10 minutes) to collect NAF-cuttings samples from each NAF-cuttings waste stream. Each NAF-cuttings sample size shall be at least one gallon. Operators shall clearly mark each container to identify each NAF-cuttings sample.
3.2 Operators shall not decant, heat, wash, or towel the NAF-cuttings to remove NAF base fluid before mass and retort analysis.
3.3 Operators shall first calculate the mass of each NAF-cuttings sample and perform the mass ratio analysis (see Equation 6, appendix 7 of subpart A of this part). Operators with only one NAF-cuttings discharge may skip this step (see Section 4.c.4, appendix 7 of subpart A of this part).
3.4 Operators shall homogenize (e.g., stirring, shaking) each NAF-cuttings sample prior to placing a sub-sample into the retort cup. The bottom of the NAF-cuttings sample container shall be examined to be sure that solids are not sticking to it.
3.5 Operators shall then calculate the NAF base fluid retained on cuttings using the retort procedure (see Equations 1-5, appendix 7 of subpart A of this part). Operators shall start the retort analyses no more than two hours after collecting the first individual mass sample for the sampling interval .
3.6 Operators shall not discharge any sample before successfully completing the mass and retort analyses [i.e., mass balance requirements (see Section 4.b, appendix 7 of subpart A of this part) are satisfied]. Operators shall immediately re-run the retort analyses if the mass balance requirements (see Equation 4, appendix 7 of subpart A of this part) are not within a tolerance of 5% (see Section 4.b, Equation 4, appendix 7 of subpart A of this part).
4.0 Calculations4.1 Operators shall calculate a set of mass percent NAF base fluid retained on cuttings values (%BF) and mass NAF-cuttings discharge fractions (X) for each NAF-cuttings waste stream (see Section 1.1 of this addendum) for each sampling interval (see Section 2.4 of this addendum) using the procedures outlined in appendix 7 of subpart A of this part.
4.2 Operators shall tabulate the following data for each individual NAF-cuttings sample: (1) Date and time of NAF-cuttings sample collection; (2) time period of NAF-cuttings sample collection (see Section 3.1 of this addendum); (3) mass and volume of each NAF-cuttings sample; (4) measured depth (feet) at NAF-cuttings sample collection (see Section 2.2 of this addendum); (5) respective linear feet of hole drilled represented by the NAF-cuttings sample (feet); and (6) the drill bit diameter (inches) used to generate the NAF-cuttings sample cuttings.
4.3 Operators shall calculate a single total mass percent NAF base fluid retained on cuttings value (%BFT) for each sampling interval (see Section 2.4 of this addendum) using the procedures outlined in appendix 7 of subpart A of this part.
4.4 Operators shall tabulate the following data for each total mass percent NAF base fluid retained on cuttings value (%BFT) for each NAF-cuttings sampling interval: (1) Date and starting and stopping times of NAF-cuttings sample collection and retort analyses; (2) measured depth of well (feet) at start of NAF-cuttings sample collection (see Section 2.2 of this addendum); (3) respective linear feet of hole drilled represented by the NAF-cuttings sample (feet); (4) the drill bit diameter (inches) used to generate the NAF-cuttings sample cuttings; and (5) annotation when zero discharge of NAF-cuttings is performed.
4.5 Operators shall calculate the weighted mass ratio averaged over all NAF well sections (%BFwell) using the procedures outlined in appendix 7 of subpart A of this part.
4.6 Operators shall tabulate the following data for each weighted mass ratio averaged over all NAF well sections (%BFwell) for each NAF well: (1) Starting and stopping dates of NAF well sections; (2) measured depth (feet) of all NAF well sections; (3) total number of sampling intervals (see Section 2.4 and Section 2.6 of this addendum); (4) number of sampling intervals tabulated during any zero discharge operations; (5) total volume of zero discharged NAF-cuttings over entire NAF well sections; and (6) identification of whether BMPs were employed (see Addendum B of appendix 7 of subpart A of this part).
Addendum B to Appendix 7 to Subpart A of Part 435 - Best Management Practices (BMPs) for use with API Recommended Practice 13B-2 1.0 Overview of BMPs1.1 Best Management Practices (BMPs) are inherently pollution prevention practices. BMPs may include the universe of pollution prevention encompassing production modifications, operational changes, material substitution, materials and water conservation, and other such measures. BMPs include methods to prevent toxic and hazardous pollutants from reaching receiving waters. Because BMPs are most effective when organized into a comprehensive facility BMP Plan, operators shall develop a BMP in accordance with the requirements in this addendum.
1.2 The BMP requirements contained in this appendix were compiled from several Regional permits, an EPA guidance document (i.e., Guidance Document for Developing Best Management Practices (BMP)” (EPA 833-B-93-004, U.S. EPA, 1993)), and draft industry BMPs. These common elements represent the appropriate mix of broad directions needed to complete a BMP Plan along with specific tasks common to all drilling operations.
1.3 Operators are not required to use BMPs if all NAF-cuttings discharges are monitored in accordance with appendix 7 of subpart A of this part.
2.0 BMP Plan Purpose and Objectives2.1 Operators shall design the BMP Plan to prevent or minimize the generation and the potential for the discharge of NAF from the facility to the waters of the United States through normal operations and ancillary activities. The operator shall establish specific objectives for the control of NAF by conducting the following evaluations.
2.2 The operator shall identify and document each NAF well that uses BMPs before starting drilling operations and the anticipated total feet to be drilled with NAF for that particular well.
2.3 Each facility component or system controlled through use of BMPs shall be examined for its NAF-waste minimization opportunities and its potential for causing a discharge of NAF to waters of the United States due to equipment failure, improper operation, natural phenomena (e.g., rain, snowfall).
2.4 For each NAF wastestream controlled through BMPs where experience indicates a reasonable potential for equipment failure (e.g., a tank overflow or leakage), natural condition (e.g., precipitation), or other circumstances to result in NAF reaching surface waters, the BMP Plan shall include a prediction of the total quantity of NAF which could be discharged from the facility as a result of each condition or circumstance.
3.0 BMP Plan Requirements3.1 The BMP Plan may reflect requirements within the pollution prevention requirements required by the Minerals Management Service (see 30 CFR 250.300) or other Federal or State requirements and incorporate any part of such plans into the BMP Plan by reference.
3.2 The operator shall certify that its BMP Plan is complete, on-site, and available upon request to EPA or the NPDES Permit controlling authority. This certification shall identify the NPDES permit number and be signed by an authorized representative of the operator. This certification shall be kept with the BMP Plan. For new or modified NPDES permits, the certification shall be made no later than the effective date of the new or modified permit. For existing NPDES permits, the certification shall be made within one year of permit issuance.
3.3 The BMP Plan shall:
3.3.1 Be documented in narrative form, and shall include any necessary plot plans, drawings or maps, and shall be developed in accordance with good engineering practices. At a minimum, the BMP Plan shall contain the planning, development and implementation, and evaluation/reevaluation components. Examples of these components are contained in “Guidance Document for Developing Best Management Practices (BMP)” (EPA 833-B-93-004, U.S. EPA, 1993).
3.3.2 Include the following provisions concerning BMP Plan review.
3.3.2.1 Be reviewed by permittee's drilling engineer and offshore installation manager (OIM) to ensure compliance with the BMP Plan purpose and objectives set forth in Section 2.0.
3.3.2.2 Include a statement that the review has been completed and that the BMP Plan fulfills the BMP Plan purpose and objectives set forth in Section 2.0. This statement shall have dated signatures from the permittee's drilling engineer and offshore installation manager and any other individuals responsible for development and implementation of the BMP Plan.
3.4 Address each component or system capable of generating or causing a release of significant amounts of NAF and identify specific preventative or remedial measures to be implemented.
4.0 BMP Plan Documentation4.1 The operator shall maintain a copy of the BMP Plan and related documentation (e.g., training certifications, summary of the monitoring results, records of NAF-equipment spills, repairs, and maintenance) at the facility and shall make the BMP Plan and related documentation available to EPA or the NPDES Permit controlling authority upon request.
5.0 BMP Plan Modification5.1 For those NAF wastestreams controlled through BMPs, the operator shall amend the BMP Plan whenever there is a change in the facility or in the operation of the facility which materially increases the generation of those NAF-wastes or their release or potential release to the receiving waters.
5.2 At a minimum the BMP Plan shall be reviewed once every five years and amended within three months if warranted. Any such changes to the BMP Plan shall be consistent with the objectives and specific requirements listed in this addendum. All changes in the BMP Plan shall be reviewed by the permittee's drilling engineer and offshore installation manager.
5.3 At any time, if the BMP Plan proves to be ineffective in achieving the general objective of preventing and minimizing the generation of NAF-wastes and their release and potential release to the receiving waters and/or the specific requirements in this addendum, the permit and/or the BMP Plan shall be subject to modification to incorporate revised BMP requirements.
6.0 Specific Pollution Prevention Requirements for NAF Discharges Associated with Cuttings6.1 The following specific pollution prevention activities are required in a BMP Plan when operators elect to control NAF discharges associated with cuttings by a set of BMPs.
6.2 Establishing programs for identifying, documenting, and repairing malfunctioning NAF equipment, tracking NAF equipment repairs, and training personnel to report and evaluate malfunctioning NAF equipment.
6.3 Establishing operating and maintenance procedures for each component in the solids control system in a manner consistent with the manufacturer's design criteria.
6.4 Using the most applicable spacers, flushes, pills, and displacement techniques in order to minimize contamination of drilling fluids when changing from water-based drilling fluids to NAF and vice versa.
6.5 A daily retort analysis shall be performed (in accordance with appendix 7 to subpart A of part 435) during the first 0.33 X feet drilled with NAF where X is the anticipated total feet to be drilled with NAF for that particular well. The retort analyses shall be documented in the well retort log. The operators shall use the calculation procedures detailed in appendix 7 to subpart A of part 435 (see Equations 1 through 8) to determine the arithmetic average (%BFwell) of the retort analyses taken during the first 0.33 X feet drilled with NAF.
6.5.1 When the arithmetic average (%BFwell) of the retort analyses taken during the first 0.33 X feet drilled with NAF is less than or equal to the base fluid retained on cuttings limitation or standard (see §§ 435.13 and 435.15), retort monitoring of cuttings may cease for that particular well. The same BMPs and drilling fluid used during the first 0.33 X feet shall be used for all remaining NAF sections for that particular well.
6.5.2 When the arithmetic average (%BFwell) of the retort analyses taken during the first 0.33 X feet drilled with NAF is greater the base fluid retained on cuttings limitation or standard (see §§ 435.13 and 435.15), retort monitoring shall continue for the following (second) 0.33 X feet drilled with NAF where X is the anticipated total feet to be drilled with NAF for that particular well. The retort analyses for the first and second 0.33 X feet shall be documented in the well retort log.
6.5.2.1 When the arithmetic average (%BFwell) of the retort analyses taken during the first 0.66 X feet (i.e., retort analyses taken from first and second 0.33 X feet) drilled with NAF is less than or equal to the base fluid retained on cuttings limitation or standard (see §§ 435.13 and 435.15), retort monitoring of cuttings may cease for that particular well. The same BMPs and drilling fluid used during the first 0.66 X feet shall be used for all remaining NAF sections for that particular well.
6.5.2.2 When the arithmetic average (%BFwell) of the retort analyses taken during the first 0.66 X feet (i.e., retort analyses taken from first and second 0.33 X feet) drilled with NAF is greater than the base fluid retained on cuttings limitation or standard (see §§ 435.13 and 435.15), retort monitoring shall continue for all remaining NAF sections for that particular well. The retort analyses for all NAF sections shall be documented in the well retort log.
6.5.3 When the arithmetic average (%BFwell) of the retort analyses taken over all NAF sections for the entire well is greater that the base fluid retained on cuttings limitation or standard (see §§ 435.13 and 435.15), the operator is in violation of the base fluid retained on cuttings limitation or standard and shall submit notification of these monitoring values in accordance with NPDES permit requirements. Additionally, the operator shall, as part of the BMP Plan, initiate a reevaluation and modification to the BMP Plan in conjunction with equipment vendors and/or industry specialists.
6.5.4 The operator shall include retort monitoring data and dates of retort-monitored and non-retort-monitored NAF-cuttings discharges managed by BMPs in their NPDES permit reports.
6.6 Establishing mud pit and equipment cleaning methods in such a way as to minimize the potential for building-up drill cuttings (including accumulated solids) in the active mud system and solids control equipment system. These cleaning methods shall include but are not limited to the following procedures.
6.6.1 Ensuring proper operation and efficiency of mud pit agitation equipment.
6.6.2 Using mud gun lines during mixing operations to provide agitation in dead spaces.
6.6.3 Pumping drilling fluids off of drill cuttings (including accumulated solids) for use, recycle, or disposal before using wash water to dislodge solids.
[66 FR 6901, Jan. 22, 2001; 66 FR 30811, June 8, 2001]Appendix 8 to Subpart A of Part 435 - Reference C16-C18 Internal Olefin Drilling Fluid Formulation
40:32.0.1.1.11.1.4.7.14 :
Appendix 8 to Subpart A of Part 435 - Reference C16-C18 Internal Olefin Drilling Fluid FormulationThe reference C16-C18 internal olefin drilling fluid used to determine the drilling fluid sediment toxicity ratio and compliance with the BAT sediment toxicity discharge limitation (see § 435.13) and NSPS (see § 435.15) shall be formulated to meet the specifications in Table 1 of this appendix.
Drilling fluid sediment toxicity ratio = 4-day LC5. of C16-C18 internal olefin drilling fluid/4-day LC5. of drilling fluid removed from drill cuttings at the solids control equipment as determined by EPA Method 1644: “Method for Conducting a Sediment Toxicity Test with Leptocheirus plumulosus and Non-Aqueous Drilling Fluids or Synthetic-Based Drilling Muds” after sediment preparation procedures specified in EPA Method 1646, which are published as appendices to Subpart A of this part and in “Analytic Methods for the Oil and Gas Extraction Point Source Category,” EPA-821-R-11-004. See § 435.11(ee) and (uu).
Table 1 - Properties for Reference C16-C18 IOs SBF Used in Discharge Sediment Toxicity Testing
| Mud weight of SBF discharged with cuttings (pounds per gallon) | Reference C16-C18 IOs SBF (pounds per gallon) | Reference C16-C18 IOs SBF synthetic to water ratio (%) |
|---|---|---|
| 8.5-11 | 9.0 | 75/25 |
| >11-14 | 11.5 | 80/20 |
| >14 | 14.5 | 85/15 |
| Plastic Viscosity (PV), centipoise (cP) | 12-30 | |
| Yield Point (YP), pounds/100 sq. ft | 10-20 | |
| 10-second gel, pounds/100 sq. ft | 8-15 | |
| 10-minute gel, pounds/100 sq. ft | 12-30 | |
| Electrical stability, V | >300 |
Appendix 1 to Subpart D of Part 435 - Procedure for Determining When Coastal Cook Inlet Operators Qualify for an Exemption From the Zero Discharge Requirement for EMO-Cuttings and SBF-Cuttings in Coastal Cook Inlet, Alaska
40:32.0.1.1.11.4.4.9.15 :
Appendix 1 to Subpart D of Part 435 - Procedure for Determining When Coastal Cook Inlet Operators Qualify for an Exemption From the Zero Discharge Requirement for EMO-Cuttings and SBF-Cuttings in Coastal Cook Inlet, Alaska 1.0 Scope and ApplicationThis appendix is to be used to determine whether a Cook Inlet, Alaska, operator in Coastal waters (Coastal Cook Inlet operator) qualifies for the exemption to the zero discharge requirement established by 40 CFR 435.43 and 435.45 for drill cuttings associated with the following non-aqueous drilling fluids: enhanced mineral oil based drilling fluids (EMO-cuttings) and synthetic-based drilling fluids (SBF-cuttings). Coastal Cook Inlet operators are prohibited from discharging oil-based drilling fluids. This appendix is intended to define those situations under which technical limitations preclude Coastal Cook Inlet operators from complying with the zero discharge requirement for EMO-cuttings and SBF-cuttings. Coastal Cook Inlet operators that qualify for this exemption may be authorized to discharge EMO-cuttings and SBF-cuttings subject to the limitations applicable to operators in Offshore waters (see subpart A of this part).
2.0 Method2.1 Any Coastal Cook Inlet operator must achieve the zero discharge limit for EMO-cuttings and SBF-cuttings unless it successfully demonstrates that technical limitations prevent it from being able to dispose of its EMO-cuttings or SBF-cuttings through on-site annular disposal, injection into a Class II underground injection control (UIC) well, or onshore land application.
2.2 To successfully demonstrate that technical limitations prevent it from being able to dispose of its EMO-cuttings or SBF-cuttings through on-site annular disposal, a Coastal Cook Inlet operator must show that it has been unable to establish formation injection in nearby wells that were initially considered for annular or dedicated disposal of EMO-cuttings or SBF-cuttings or prove to the satisfaction of the Alaska Oil and Gas Conservation Commission (AOGCC) that the EMO-cuttings or SBF-cuttings will be confined to the formation disposal interval. This demonstration must include:
a. Documentation, including engineering analysis, that shows (1) an inability to establish formation injection (e.g., formation is too tight), (2) an inability to confine EMO-cuttings or SBF-cuttings in disposal formation (e.g., no confining zone or adequate barrier to confine wastes in formation), or (3) the occurrence of high risk emergency (e.g., mechanical failure of well, loss of ability to inject that risks loss of well which would cause significant economic harm or create a substantial risk to safety); and
b. A risk analysis of alternative disposal options, including environmental assessment, human health and safety, and economic impact, that shows discharge as the lowest risk option.
2.3 To successfully demonstrate that technical limitations prevent it from being able to dispose of its EMO-cuttings or SBF-cuttings through injection into a Class II UIC well, a Coastal Cook Inlet operator must show that it has been unable to establish injection into a Class II UIC well or prove to the satisfaction of the Alaska Oil and Gas Conservation Commission (AOGCC) that the EMO-cuttings or SBF-cuttings will be confined to the formation disposal interval. This demonstration must include:
a. Documentation, including engineering analysis, that shows the inability to confine EMO-cuttings or SBF-cuttings in a Class II UIC well (e.g., no confining zone or adequate barrier to confine wastes in formation);
b. Documentation demonstrating that no Class II UIC well is accessible (e.g., operator does not own, competitor will not allow injection); and
c. A risk analysis of alternative disposal option, including environmental assessment, human health and safety, and economic impact, that shows discharge as the lowest risk option.
2.4 To successfully demonstrate that technical limitations prevent it from being able to dispose of its EMO-cuttings or SBF-cuttings through land application, a Coastal Cook Inlet operator must show that it has been unable to handle drilling waste or dispose of EMO-cuttings or SBF-cuttings at an appropriate land disposal site. This demonstration must include:
a. Documentation of site restrictions that preclude land application (e.g., no land disposal sites available);
b. Documentation of the platform's lack of capacity for adequate storage of EMO-cuttings or SBF-cuttings (e.g., limited storage or room for cuttings transfer); or
c. Documentation of inability to transfer EMO-cuttings or SBF-cuttings from platform to land for disposal (e.g., extremely low tides, high wave action).
3.0 Procedure3.1 Except as described in Section 3.2 of this appendix, a Coastal Cook Inlet operator believing that it qualifies for the exemption to the zero discharge requirement for EMO-cuttings or SBF-cuttings must apply for and obtain an individual NPDES permit prior to discharging EMO-cuttings or SBF-cuttings to waters of the United States.
3.2 Discharges occurring as the result a high risk emergency (e.g., mechanical failure of well, loss of ability to inject that risks loss of well which would cause significant economic harm or safety) may be authorized by a general NPDES permit provided that:
a. The Coastal Cook Inlet operator satisfactorily demonstrates to EPA Region 10 the fulfillment of the other exemption requirements described in Section 2.0 of this appendix, or
b. The general permit allows for high risk emergency discharges and provides Reporting Requirements to EPA Region 10 immediately upon commencing discharge.
[66 FR 6918, Jan. 22, 2001]