Title 40
PART 430 APPENDIX A
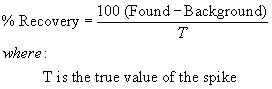
| Paper or pulp mill stream | Sample volume (mL)* |
Adsorption volume (mL) |
|---|---|---|
| Evaporator condensate | 100.0 | 100 |
| Process water | 100.0 | 100 |
| Pulp mill effluent | 30.0 | 50 |
| Paper mill effluent | 10.0 | 25 |
| Combined mill effluent | 5.0 | 25 |
| Combined bleach effluent | 1.0 | 25 |
| C-stage filtrate | 0.5 | 25 |
| E-stage filtrate | 0.5 | 25 |
* Assumes dilution to final volume of 100 mL. All sample aliquots (replicates, diluted samples) must be analyzed using the same fixed final volume (sample volume plus reagent water, as needed).
11.1.2 Sample dilution procedure.
11.1.2.1 Partially fill a precleaned volumetric flask with pH <2 reagent water, allowing for the volume of sample to be added.
11.1.2.2 Mix sample thoroughly by tumbling or shaking vigorously.
11.1.2.3 Immediately withdraw the required sample aliquot using a pipet or micro-syringe.
Note:Because it will be necessary to rinse the pipet or micro-syringe (Section 11.1.2.5), it may be necessary to pre-calibrate the pipet or micro-syringe to assure that the exact volume desired will be delivered.
11.1.2.4 Dispense or inject the aliquot into the volumetric flask.
11.1.2.5 Rinse the pipet or syringe with small portions of reagent water and add to the flask.
11.1.2.6 Dilute to the mark with pH <2 reagent water.
11.1.3 All samples to be reported for regulatory compliance monitoring purposes must be analyzed in duplicate, as described in Section 11.5.
11.1.4 Pulp and Paper in-process samples: The concentration of organic halide in in-process samples has been shown to be 20 to 30% greater using the micro-column adsorption technique than using the batch adsorption technique. For this reason, the micro-column technique shall be used for monitoring in-process samples. Examples of in-process samples include: combined bleach plant effluent, C-stage filtrate, and E-stage filtrate.
11.2 Batch adsorption and filtration.
11.2.1 Place the appropriate volume of sample (diluted if necessary), preserved as described in Section 8, into an Erlenmeyer flask.
11.2.2 Add 5 mL of nitrate stock solution to the sample aliquot.
11.2.3 Add one level scoop of activated carbon that has passed the quality control tests in Section 9.
11.2.4 Shake the suspension for at least one hour in a mechanical shaker.
11.2.5 Filter the suspension through a polycarbonate membrane filter. Filter by suction until the liquid level reaches the top of the carbon.
11.2.6 Wash the inside surface of the filter funnel with 25 mL (±5 mL) of nitrate wash solution in several portions. After the level of the final wash reaches the top of the GAC, filter by suction until the cake is barely dry. The time required for drying should be minimized to prevent exposure of the GAC to halogen vapors in the air, but should be sufficient to permit drying of the cake so that excess water is not introduced into the combustion apparatus. A drying time of approximately 10 seconds under vacuum has been shown to be effective for this operation.
11.2.7 Carefully remove the top of the filter holder, making sure that no carbon is lost. This operation is most successfully performed by removing the clamp, tilting the top of the filter holder (the funnel portion) to one side, and lifting upward.
11.2.8 Using a squeeze bottle or micro-syringe, rapidly rinse the carbon from the inside of the filter holder onto the filter cake using small portions of wash solution. Allow the cake to dry under vacuum for no more than 10 seconds after the final rinse. Immediately turn the vacuum off.
11.2.9 Using tweezers, carefully fold the polycarbonate filter in half, then in fourths, making sure that no carbon is lost.
11.3 Column adsorption.
11.3.1 Column preparation: Prepare a sufficient number of columns for one day's operation as follows:
11.3.1.1 In a glove box or area free from halide vapors, place a plug of Cerafelt into the end of a clean glass column.
11.3.1.2 Fill the glass column with one level scoop (approximately 40 mg) of granular activated carbon that has passed the quality control tests in Section 9.
11.3.1.3 Insert a Cerafelt plug into the open end of the column to hold the carbon in place.
11.3.1.4 Store the columns in a glass jar with PTFE lined screw-cap to prevent infiltration of halide vapors from the air.
11.3.2 Column setup.
11.3.2.1 Install two columns in series in the adsorption module.
11.3.2.2 If the sample is known or expected to contain particulates that could prevent free flow of sample through the micro-columns, a Cerafelt plug is placed in the tubing ahead of the columns. If a measurement of the OX content of the particulates is desired, the Cerafelt plug can be washed with nitrate solution, placed in a combustion boat, and processed as a separate sample.
11.3.3 Adjusting sample flow rate: Because the flow rate used to load the sample onto the columns can affect the ability of the GAC to adsorb organic halides, the flow rate of the method blank is measured, and the gas pressure used to process samples is adjusted accordingly. The flow rate of the blank, which is composed of acidified reagent water and contains no particulate matter, should be greater than the flow rate of any sample containing even small amounts of particulate matter.
11.3.3.1 Fill the sample reservoir with the volume of reagent water chosen for the analysis (Section 9.4.1.2) that has been preserved and acidified as described in Section 8. Cap the reservoir.
11.3.3.2 Adjust the gas pressure per the manufacturer's instructions. Record the time required for the entire volume of reagent water to pass through both columns. The flow rate must not exceed 3 mL/min over the duration of the time required to adsorb the volume. If this flow rate is exceeded, adjust gas pressure, prepare another blank, and repeat the adsorption.
11.3.3.3 Once the flow rate for the blank has been established, the same adsorption conditions must be applied to all subsequent samples during that eight-hour shift, or until another method blank is processed, whichever comes first. To aid in overcoming breakthrough problems, a lower gas pressure (and, therefore, flow rate) may be used for processing of samples, if desired. If the sample adsorption unit is disassembled or cleaned, the flow rate must be checked before processing additional samples.
11.3.3.4 Elute the pair of columns with 2 mL of nitrate wash solution. The flow rate of nitrate wash solution must not exceed 3 mL/min.
11.3.3.5 Separate the columns and mark for subsequent analysis.
11.3.4 The adsorption of sample volumes is performed in a similar fashion. Fill the sample reservoir with the sample volume chosen for the analysis (Section 11.1), that has been preserved as described in Section 8. All analyses must be performed with this volume (sample volume plus reagent water, as needed) in order to maintain a flow rate no greater than that determined for the blank (see Section 11.3.3).
11.3.4.1 Use the same gas pressure for sample adsorption as is used for the blank.
11.3.4.2 Elute the columns with 2 mL of the nitrate wash solution.
11.3.4.3 Separate the columns and mark for subsequent analysis.
11.3.5 If it is desirable to make measurements at levels lower than can be achieved with the sample volume chosen, or if the instrument response of an undiluted sample is less than three times the instrument response of the blank (Section 12.6.3), a larger sample volume must be used.
11.4 Combustion and titration.
11.4.1 Polycarbonate filter and GAC from batch adsorption.
11.4.1.1 Place the folded polycarbonate filter containing the GAC in a quartz combustion boat, close the airlock, and proceed with the automated sequence.
11.4.1.2 Record the signal from the micro-coulometer for a minimum integration time of 10 minutes and determine the concentration of Cl− from calibration data, per Section 12.
11.4.2 Columns from column adsorption.
11.4.2.1 Using the push rod, push the carbon and the Cerafelt plug(s) from the first column into a combustion boat. Proceed with the automated sequence.
11.4.2.2 Record the signal from the micro-coulometer for a minimum integration time of 10 minutes and determine the concentration of Cl− for the first column from calibration data, per Section 12.
11.4.2.3 Repeat the automated sequence with the second column.
11.4.2.4 Determine the extent of breakthrough of organic halides from the first column to the second column, as described in Section 12.
11.4.3 The two columns that are used for the method blank must be combusted separately, as is done for samples. 11.5 Duplicate sample analysis: All samples to be reported for regulatory compliance purposes must be analyzed in duplicate. This requirement applies to both the batch and column adsorption procedures. In addition, if it is necessary to dilute the sample for the purposes of reducing breakthrough or maintaining the concentration within the calibration range, a more or less dilute sample must be analyzed. The adsorption volumes used for analysis of undiluted samples, diluted samples, and all replicates must be the same as the volume used for QC tests and calibration (Sections 9 and 10).
11.5.1 Using results from analysis of one sample volume (Section 11.4) and the procedure in Section 11.1.2, determine if the dilution used was within the calibration range of the instrument and/or if breakthrough exceeded the specification in Section 12.3.1. If the breakthrough criterion was exceeded or the sample was not within the calibration range, adjust the dilution volume as needed. If the breakthrough criterion was not exceeded and the sample dilution was within the calibration range, a second volume at the same dilution level may be used.
11.5.2 Adsorb the sample using the same technique (batch or column) used for the first sample volume. Combust the GAC from the second volume as described in Section 11.4, and calculate the results as described in Section 12. Compare the results of the two analyses as described in Section 12.4.
11.5.3 Duplicate analyses are not required for method blanks, as different dilution levels are not possible.
11.5.4 Duplicate analyses of the PAR standard used for calibration verification (Section 9.10) are not required.
12.0 Data Analysis and Calculations12.1 Batch Adsorption Method: Calculate the blank-subtracted concentration of adsorbable organic halide detected in each sample (in micrograms of chloride per liter) using the following equation:
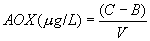 Where:
C=µg Cl− from micro-coulometer for the sample B=µg Cl− from
micro-coulometer for the reagent water blank (Section 9.4.1) V =
volume of sample in liters
Where:
C=µg Cl− from micro-coulometer for the sample B=µg Cl− from
micro-coulometer for the reagent water blank (Section 9.4.1) V =
volume of sample in liters
This calculation is performed for each of the two dilution levels analyzed for each sample.
12.2 Column Adsorption Method: Calculate the blank-subtracted concentration of adsorbable organic halide detected in each sample (in micrograms of chloride per liter) using the following equation:
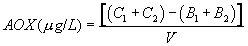 C1=µg Cl−
from micro-coulometer for first column from the sample C2=µg Cl−
from micro-coulometer for second column from the sample B1=µg from
micro-coulometer for first column from the reagent water blank
(Section 9.4.1) B2=µg Cl− from micro-coulometer for second column
from the reagent water blank (Section 9.4.1) V = volume of sample
in liters
C1=µg Cl−
from micro-coulometer for first column from the sample C2=µg Cl−
from micro-coulometer for second column from the sample B1=µg from
micro-coulometer for first column from the reagent water blank
(Section 9.4.1) B2=µg Cl− from micro-coulometer for second column
from the reagent water blank (Section 9.4.1) V = volume of sample
in liters
12.3 Percent breakthrough: For each sample analyzed by the column method, calculate the percent breakthrough of halide from the first column to the second column, using the following equation:
12.3.1 For samples to be reported for regulatory compliance purposes, the percent breakthrough must be less than or equal to 25% for both of the two analyses performed on each sample (see Section 11.5).
12.3.2 If the breakthrough exceeds 25%, dilute the affected sample further, maintaining the amount of halide at least three times higher than the level of blank, and reanalyze the sample. Ensure that the sample is also analyzed at a second level of dilution that is at least a factor of 2 different (and still higher than three times the blank).
12.4 Relative percent difference (RPD): Calculate the relative percent difference between the results of the two analyses of each sample, using the following equation:
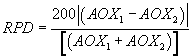
12.5 High concentrations of AOX: If the amount of halide from either analysis exceeds the calibration range, dilute the sample and reanalyze, maintaining at least a factor of 2 difference in the dilution levels of the two portions of the sample used.
12.6 Low concentrations of AOX: The blank-subtracted final result from the batch procedure or the sum of the blank-subtracted results from the two carbon columns should be significantly above the level of the blank.
12.6.1 If the instrument response for a sample exceeds the instrument response for the blank by a factor of at least 3, the result is acceptable.
12.6.2 If the instrument response for a sample is less than three times the instrument response for the blank, and the sample has been diluted, analyze a less dilute aliquot of sample.
12.6.3 If the instrument response of an undiluted sample containing AOX above the minimum level is less than three times the instrument response for the blank, the result is suspect and may not be used for regulatory compliance purposes. In this case, find the cause of contamination, correct the problem, and reanalyze the sample under the corrected conditions.
12.7 Report results that meet all of the specifications in this method as the mean of the blank-subtracted values from Section 12.1 or 12.2 for the two analyses at different dilution levels, in µg/L of Cl− (not as 2,4,6-trichlorophenol), to three significant figures. Report the RPD of the two analyses. For samples analyzed by the column procedure, also report the percent breakthrough.
13.0 Method PerformanceThe specifications contained in this method are based on data from a single laboratory and from a large-scale study of the pulp and paper industry.
14.0 Pollution Prevention14.1 The solvents used in this method pose little threat to the environment when recycled and managed properly.
14.2 Standards should be prepared in volumes consistent with laboratory use to minimize the volume of expired standards to be disposed.
15.0 Waste Management15.1 It is the laboratory's responsibility to comply with all federal, state, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions, and to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance with all sewage discharge permits and regulations is also required.
15.2 Samples preserved with HCl or H2SO4 to pH <2 are hazardous and must be neutralized before being disposed, or must be handled as hazardous waste. Acetic acid and silver acetate solutions resulting from cell flushing must be disposed of in accordance with all applicable federal, state, and local regulations.
15.3 For further information on waste management, consult “The Waste Management Manual for Laboratory Personnel,” and “Less is Better: Laboratory Chemical Management for Waste Reduction,” both available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street NW., Washington, DC 20036.
16.0 References16.1 “Total Organic Halide, Methods 450.1 - Interim,” Prepared by Stephen Billets and James J. Lichtenberg, USEPA, Office of Research and Development, Physical and Chemical Methods Branch, EMSL-Cincinnati, Cincinnati, OH 45268, EPA 600/4-81-056 (1981).
16.2 Method 9020, USEPA Office of Solid Waste, “Test Methods for Evaluating Solid Waste, SW-846,” Third Edition, 1987.
16.3 “Determination of Adsorbable Organic Halogens (AOX),” “German Standard Methods for the Analysis of Water, Waste Water and Sludge - General Parameters of Effects and Substances,” Deutsche Industrie Norm (DIN) Method 38 409, Part 14, DIN German Standards Institute, Beuth Verlag, Berlin, Germany (1987).
16.4 “Water Quality: Determination of Adsorbable Organic Halogens (AOX),” International Organization for Standard/Draft International Standardization (ISO/DIS) Method 9562 (1988).
16.5 “Organically Bound Chlorine by the AOX Method,” SCAN-W 9:89, Secretariat, Scandinavian Pulp, Paper and Board Testing Committee, Box 5604, S-11486, Stockholm, Sweden (1989).
16.6 Method 5320, “Dissolved Organic Halogen,” from “Standard Methods for the Examination of Water and Wastewater,” 5320, American Public Health Association, 1015 15th St. NW, Washington, DC 20005 (1989).
16.7 “Canadian Standard Method for the Determination of Adsorbable Organic Halides (AOX) in Waters and Wastewaters,” Environment Canada and The Canadian Pulp and Paper Association (1990).
16.8 40 CFR part 136, appendix B.
16.9 “Working with Carcinogens,” DHEW, PHS, CDC, NIOSH, Publication 77-206, (Aug 1977).
16.10 “OSHA Safety and Health Standards, General Industry” OSHA 2206, 29 CFR 1910 (Jan 1976).
16.11 “Safety in Academic Chemistry Laboratories,” ACS Committee on Chemical Safety (1979).
16.12 “Methods 330.4 and 330.5 for Total Residual Chlorine,” USEPA, EMSL-Cincinnati, Cincinnati, OH 45268, EPA-4-79-020 (March 1979).
16.13 “Validation of Method 1650: Determination of Organic Halide,” Analytical Technologies Inc., ERCE Contract 87-3410, November 15, 1990. Available from the EPA Sample Control Center, DynCorp, 300 N. Lee St., Alexandria, VA 22314 (703-519-1140).
17.0 Figures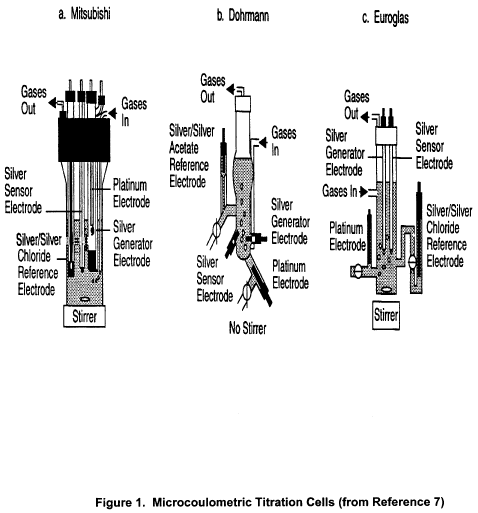
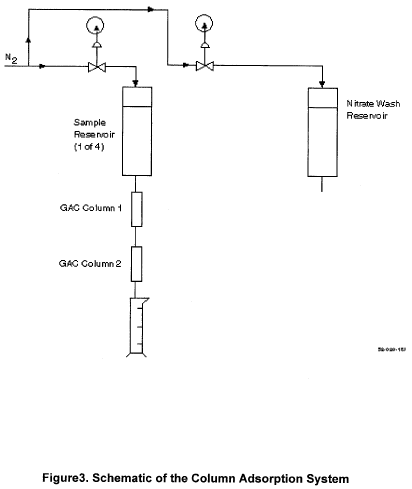
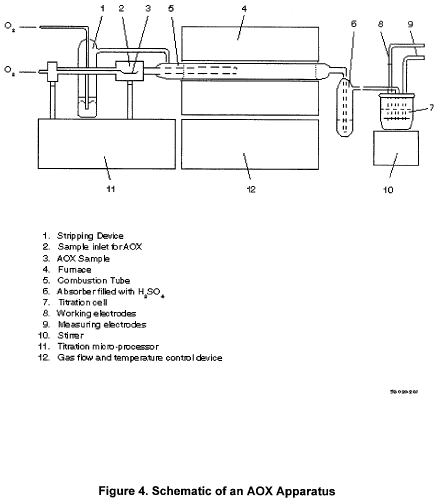 18.0 Glossary of
Definitions and Purposes
18.0 Glossary of
Definitions and Purposes
These definitions and purposes are specific to this method but have been conformed to common usage as much as possible.
18.1 Units of weight and measure and their abbreviations.
18.1.1 Symbols.
°C degrees Celsius µg microgram µL microliter < less than > greater than % percent18.1.2 Alphabetical characters.
cm centimeter g gram h hour ID inside diameter in inch L liter m meter mg milligram min minute mL milliliter mm millimeter N normal; gram molecular weight of solute divided by hydrogen equivalent of solute, per liter of solution OD outside diameter ppb part-per-billion ppm part-per-million ppt part-per-trillion psig pounds-per-square inch gauge v/v volume per unit volume w/v weight per unit volume18.2 Definitions and acronyms (in alphabetical order).
Analyte: AOX tested for by this method.
Calibration standard (CAL): A solution prepared from a secondary standard and/or stock solution which is used to calibrate the response of the instrument with respect to analyte concentration.
Calibration verification standard (VER): The mid-point calibration standard (CS3) that is used to verify calibration.
Field blank: An aliquot of reagent water or other reference matrix that is placed in a sample container in the laboratory or the field, and treated as a sample in all respects, including exposure to sampling site conditions, storage, preservation, and all analytical procedures. The purpose of the field blank is to determine if the field or sample transporting procedures and environments have contaminated the sample.
IPR: Initial precision and recovery; four aliquots of the diluted PAR standard analyzed to establish the ability to generate acceptable precision and accuracy. An IPR is performed prior to the first time this method is used and any time the method or instrumentation is modified.
Laboratory blank: See Method blank.
Laboratory control sample (LCS): See Ongoing precision and recovery sample (OPR).
Laboratory reagent blank: See Method blank.
May: This action, activity, or procedural step is neither required nor prohibited.
May not: This action, activity, or procedural step is prohibited.
Method blank: An aliquot of reagent water that is treated exactly as a sample including exposure to all glassware, equipment, solvents, reagents, internal standards, and surrogates that are used with samples. The method blank is used to determine if analytes or interferences are present in the laboratory environment, the reagents, or the apparatus.
Minimum level (ML): The level at which the entire analytical system must give a recognizable signal and acceptable calibration point for the analyte. It is equivalent to the concentration of the lowest calibration standard, assuming that all method-specified sample weights, volumes, and cleanup procedures have been employed.
Must: This action, activity, or procedural step is required.
OPR: Ongoing precision and recovery standard; a laboratory blank spiked with a known quantity of analyte. The OPR is analyzed exactly like a sample. Its purpose is to assure that the results produced by the laboratory remain within the limits specified in this method for precision and recovery.
PAR: Precision and recovery standard; secondary standard that is diluted and spiked to form the IPR and OPR.
Preparation blank: See Method blank.
Primary dilution standard: A solution containing the specified analytes that is purchased or prepared from stock solutions and diluted as needed to prepare calibration solutions and other solutions.
Quality control check sample (QCS): A sample containing all or a subset of the analytes at known concentrations. The QCS is obtained from a source external to the laboratory or is prepared from a source of standards different from the source of calibration standards. It is used to check laboratory performance with test materials prepared external to the normal preparation process.
Reagent water: Water demonstrated to be free from the analyte of interest and potentially interfering substances at the method detection limit for the analyte.
Relative standard deviation (RSD): The standard deviation multiplied by 100, divided by the mean.
RSD: See Relative standard deviation.
Should: This action, activity, or procedural step is suggested but not required.
Stock solution: A solution containing an analyte that is prepared using a reference material traceable to EPA, the National Institute of Science and Technology (NIST), or a source that will attest to the purity and authenticity of the reference material.
VER: See Calibration verification standard.
Method 1653 - Chlorinated Phenolics in Wastewater by In Situ Acetylation and GCMS 1.0 Scope and Application1.1 This method is for determination of chlorinated phenolics (chlorinated phenols, guaiacols, catechols, vanillins, syringaldehydes) and other compounds associated with the Clean Water Act; the Resource Conservation and Recovery Act; and the Comprehensive Environmental Response, Compensation, and Liability Act; and that are amenable to in situ acetylation, extraction, and analysis by capillary column gas chromatography/mass spectrometry (GCMS). This method is based on existing methods for determination of chlorophenolics in pulp and paper industry wastewaters (References 1 and 2).
1.2 The chemical compounds listed in Table 1 may be determined in waters and, specifically, in in-process streams and wastewaters associated with the pulp and paper industry. The method is designed to meet the survey and monitoring requirements of the Environmental Protection Agency (EPA).
1.3 The detection limit of this method is usually dependent on the level of interferences rather than instrumental limitations. The method detection limits (MDLs) in Table 2 typify the minimum quantity that can be detected with no interferences present.
1.4 The GCMS portions of this method are for use only by persons experienced with GCMS or under the close supervision of such qualified persons. Laboratories unfamiliar with analyses of environmental samples by GCMS should run the performance tests in Reference 3 before beginning.
1.5 Any modification of the method beyond those expressly permitted is subject to the application and approval of alternative test procedures under 40 CFR parts 136.4 and 136.5.
2.0 Summary of Method2.1 A 1000-mL aliquot of water is spiked with stable isotopically labeled analogs of the compounds of interest and an internal standard. The solution is adjusted to neutral pH, potassium carbonate buffer is added, and the pH is raised to 9-11.5. The chlorophenolics are converted in situ to acetates by the addition of acetic anhydride. After acetylation, the solution is extracted with hexane. The hexane is concentrated to a final volume of 0.5 mL, an instrument internal standard is added, and an aliquot of the concentrated extract is injected into the gas chromatograph (GC). The compounds are separated by GC and detected by a mass spectrometer (MS). The labeled compounds and internal standard serve to correct the variability of the analytical technique.
2.2 Identification of a pollutant (qualitative analysis) is performed by comparing the relative retention time and mass spectrum to that of an authentic standard. A compound is identified when its relative retention time and mass spectrum agree.
2.3 Quantitative analysis is performed in one of two ways by GCMS using extracted ion-current profile (EICP) areas: (1) For those compounds listed in Table 1 for which standards and labeled analogs are available, the GCMS system is calibrated and the compound concentration is determined using an isotope dilution technique; (2) for those compounds listed in Table 1 for which authentic standards but no labeled compounds are available, the GCMS system is calibrated and the compound concentration is determined using an internal standard technique.
2.4 Quality is assured through reproducible calibration and testing of the extraction and GCMS systems.
3.0 Definitions3.1 Chlorinated phenolics are the chlorinated phenols, guaiacols, catechols, vanillins, syringaldehydes and other compounds amenable to in situ acetylation, extraction, and determination by GCMS using this method.
3.2 Definitions for other terms used in this method are given in the glossary at the end of the method (Section 20.0).
4.0 Interferences4.1 Solvents, reagents, glassware, and other sample processing hardware may yield artifacts and/or elevated baselines, causing misinterpretation of chromatograms and spectra. All materials used in the analysis shall be demonstrated to be free from interferences under the conditions of analysis by running method blanks initially and with each sample batch (samples started through the extraction process on a given eight-hour shift, to a maximum of 20). Specific selection of reagents and purification of solvents by distillation in all-glass systems may be required. Glassware and, where possible, reagents are cleaned by using solvent rinse and baking at 450 °C for a minimum of one hour.
4.2 Interferences co-extracted from samples will vary considerably from source to source, depending on the diversity of the site being sampled. Industry experience suggests that high levels of non-chlorinated phenols may cause poor recovery of the compounds of interest, particularly in samples collected in the vicinity of a source of creosote, such as a wood-preserving plant (Reference 1).
4.3 The internal standard, 3,4,5-trichlorophenol, has been reported to be an anaerobic degradation product of 2,3,4,5-tetrachlorophenol and/or pentachlorophenol (Reference 1). When an interference with this or another compound occurs, labeled pentachlorophenol or another labeled compound may be used as an alternative internal standard; otherwise, the internal standards and reference compounds must be used as specified in this method.
4.4 Blank contamination by pentachlorophenol has been reported (Reference 1) to be traceable to potassium carbonate; it has also been reported that this contamination may be removed by baking overnight at 400 to 500 °C.
4.5 Catechols are susceptible to degradation by active sites on injection port liners and columns, and are subject to oxidation to the corresponding chloro-o-benzoquinones (Reference 2). A small amount of ascorbic acid may be added to samples to prevent auto-oxidation (Reference 2; also see Section 11.1.6). For pulp and paper industry samples, ascorbic acid may be added to treated effluent samples only.
5.0 Safety5.1 The toxicity or carcinogenicity of each compound or reagent used in this method has not been precisely determined; however, each chemical compound should be treated as a potential health hazard. Exposure to these compounds should be reduced to the lowest possible level. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of materials safety data sheets (MSDSs) should be made available to all personnel involved in these analyses. Additional information on laboratory safety can be found in References 4 through 6.
5.2 Samples may contain high concentrations of toxic compounds, and should be handled with gloves and a hood opened to prevent exposure.
6.0 Equipment and Supplies Note:Brand names, suppliers, and part numbers are for illustrative purposes only. No endorsement is implied. Equivalent performance may be achieved using apparatus and materials other than those specified here, but demonstration of equivalent performance that meets the requirements of this method is the responsibility of the laboratory.
6.1 Sampling equipment for discrete or composite sampling.
6.1.1 Sample bottles and caps.
6.1.1.1 Sample bottle: Amber glass, 1000-mL minimum, with screw-cap. If amber bottles are not available, samples shall be protected from light.
6.1.1.2 Bottle caps: Threaded to fit sample bottles. Caps shall be lined with PTFE.
6.1.1.3 Cleaning bottles: Detergent water wash, cap with aluminum foil, and bake at 450 °C for a minimum of one hour before use.
6.1.1.4 Cleaning liners: Detergent water wash, reagent water (Section 7.4) and solvent rinse, and bake at approximately 200 °C for a minimum of 1 hour prior to use.
6.1.1.5 Bottles and liners must be lot-certified to be free of chlorophenolics by running blanks according to this method. If blanks from bottles and/or liners without cleaning or with fewer cleaning steps show no detectable chlorophenolics, the bottle and liner cleaning steps that do not eliminate chlorophenolics may be omitted.
6.1.2 Compositing equipment: Automatic or manual compositing system incorporating glass containers cleaned per bottle cleaning procedure above. Sample containers are kept at 0 to 4 °C during sampling. Glass or PTFE tubing only shall be used. If the sampler uses a peristaltic pump, a minimum length of compressible silicone rubber tubing may be used in the pump only. Before use, the tubing shall be thoroughly rinsed with methanol, followed by repeated rinsing with reagent water (Section 7.4) to minimize sample contamination. An integrating flow meter is used to collect proportional composite samples.
6.2 Extraction apparatus.
6.2.1 Bottle or beaker: 1500-to 2000-mL capacity.
6.2.2 Separatory funnel: 500-to 2000-mL, glass, with PTFE stopcock.
6.2.3 Magnetic stirrer: Corning Model 320, or equivalent, with stirring bar.
6.3 Polyethylene gloves: For handling samples and extraction equipment (Fisher 11-394-110-B, or equivalent).
6.4 Graduated cylinders: 1000-mL, 100-mL, and 10-mL nominal.
6.5 Centrifuge: Capable of accepting 50-mL centrifuge tubes and achieving 3000 RPM.
6.5.1 Centrifuge tubes.
6.5.1.1 35-mL nominal, with PTFE-lined screw-cap.
6.5.1.2 15-mL nominal, conical graduated, with ground-glass stopper.
6.6 Concentration apparatus.
6.6.1 Kuderna-Danish (K-D) concentrator tube: 10-mL, graduated (Kontes K-570050-1025, or equivalent) with calibration verified. Ground-glass stopper (size 19/22 joint) is used to prevent evaporation of extracts.
6.6.2 Kuderna-Danish (K-D) evaporation flask: 1000-mL (Kontes K-570001-1000, or equivalent), attached to concentrator tube with springs (Kontes K-662750-0012).
6.6.3 Snyder column: Three-ball macro (Kontes K-503000-0232, or equivalent).
6.6.4 Snyder column: Two-ball micro (Kontes K-469002-0219, or equivalent).
6.6.5 Boiling chips: Approximately 10/40 mesh, extracted with methylene chloride and baked at 450 °C for a minimum of one hour.
6.6.6 Nitrogen evaporation apparatus: Equipped with a water bath controlled at 35 to 40 °C (N-Evap, Organomation Associates, Inc., South Berlin, MA, or equivalent), installed in a fume hood. This device may be used in place of the micro-Snyder column concentrator in Section 6.6.4 above.
6.7 Water bath: Heated, with concentric ring cover, capable of temperature control (±2 °C), installed in a fume hood.
6.8 Sample vials: Amber glass, 1- to 3-mL, with PTFE-lined screw-cap.
6.9 Balances.
6.9.1 Analytical: Capable of weighing 0.1 mg.
6.9.2 Top loading: Capable of weighing 10 mg.
6.10 pH meter.
6.11 Gas chromatograph: Shall have splitless or on-column injection port for capillary column, temperature program with 50 °C hold, and shall meet all of the performance specifications in Section 9.
6.12 Gas chromatographic column: 30 m (±5 m) × 0.25 mm (±0.02 mm) I.D. × 0.25 micron, 5% phenyl, 94% methyl, 1% vinyl silicone bonded-phase fused-silica capillary column (J & W DB-5, or equivalent).
6.13 Mass spectrometer: 70 eV electron impact ionization, shall repetitively scan from 42 to 450 amu in 0.95 to 1.00 second, and shall produce a unit resolution (valleys between m/z 441-442 less than 10% of the height of the 441 peak), background-corrected mass spectrum from 50 ng decafluorotriphenylphosphine (DFTPP) introduced through the GC inlet. The spectrum shall meet the mass-intensity criteria in Table 3 (Reference 7). The mass spectrometer shall be interfaced to the GC such that the end of the capillary column terminates within 1 cm of the ion source, but does not intercept the electron or ion beams. All portions of the column which connect the GC to the ion source shall remain at or above the column temperature during analysis to preclude condensation of less volatile compounds.
6.14 Data system: Shall collect and record MS data, store mass-intensity data in spectral libraries, process GCMS data, generate reports, and compute and record response factors.
6.14.1 Data acquisition: Mass spectra shall be collected continuously throughout the analysis and stored on a mass storage device.
6.14.2 Mass spectral libraries: User-created libraries containing mass spectra obtained from analysis of authentic standards shall be employed to reverse search GCMS runs for the compounds of interest (Section 10.2).
6.14.3 Data processing: The data system shall be used to search, locate, identify, and quantify the compounds of interest in each GCMS analysis. Software routines shall be employed to compute retention times, and to compute peak areas at the m/z's specified (Table 4). Displays of spectra, mass chromatograms, and library comparisons are required to verify results.
6.14.4 Response factors and multi-point calibrations: The data system shall be used to record and maintain lists of response factors (response ratios for isotope dilution) and multi-point calibration curves (Section 10). Computations of relative standard deviation (coefficient of variation) are used for testing calibration linearity. Statistics on initial (Section 9.3.2) and ongoing (Section 9.6) performance shall be computed and maintained.
7.0 Reagents and Standards7.1 Reagents for adjusting sample pH.
7.1.1 Sodium hydroxide: Reagent grade, 6 N in reagent water.
7.1.2 Sulfuric acid: Reagent grade, 6 N in reagent water.
7.2 Reagents for sample preservation.
7.2.1 Sodium thiosulfate (Na2S2O3) solution (1 N): Weigh 79 g Na2S2O3 in a 1-L volumetric flask and dilute to the mark with reagent water.
7.2.2 Ascorbic acid solution: Prepare a solution of ascorbic acid in reagent water at a concentration of 0.1 g/mL. This solution must be prepared fresh on each day when derivatizations will be performed. Therefore, do not prepare more than will be used that day. (A 50-mL volume is sufficient for ten analyses).
7.3 Solvents: Hexane, acetone, and methanol. Distilled in glass (Burdick and Jackson, or equivalent).
7.4 Reagent water: Water in which the compounds of interest and interfering compounds are not detected by this method.
7.5 Reagents for derivatization.
7.5.1 Potassium carbonate (K2CO3).
7.5.1.1 Purification: Spread in a shallow baking dish, heat overnight at 400 to 500 °C.
7.5.1.2 Solution: Dissolve 150 g purified K2CO3 in 250 mL reagent water.
7.5.2 Acetic anhydride: Redistilled reagent grade.
7.6 Analytical standards.
7.6.1 Derivatization: Because the chlorinated phenolics are determined as their acetate derivatives after in situ acetylation, the method requires that the calibration standards be prepared by spiking the underivatized materials into reagent water and carrying the spiked reagent water aliquot through the entire derivatization and extraction procedure that is applied to the field samples.
7.6.2 Standard solutions: Purchased as solutions or mixtures with certification to their purity, concentration, and authenticity, or prepared from materials of known purity and composition. If chemical purity of a compound is 98% or greater, the weight may be used without correction to compute the concentration of the standard. When not being used, standards are stored in the dark at −20 to −10 °C in screw-capped vials with PTFE-lined lids. A mark is placed on the vial at the level of the solution so that solvent evaporation loss can be detected. The vials are brought to room temperature prior to use.
7.6.3 If the chemical purity of any standard does not meet the 98% requirement above, the laboratory must correct all calculations, calibrations, etc., for the difference in purity.
7.7 Preparation of stock solutions: Prepare chlorovanillins and chlorosyringaldehydes in acetone, as these compounds are subject to degradation in methanol. Prepare the remaining chlorophenolics in methanol. Prepare all standards per the steps below. Observe the safety precautions in Section 5.
7.7.1 Dissolve an appropriate amount of assayed reference material in a suitable solvent. For example, weigh 50 mg (±0.1 mg) of pentachlorophenol in a 10-mL ground-glass-stoppered volumetric flask and fill to the mark with methanol. After the pentachlorophenol is completely dissolved, transfer the solution to a 15-mL vial with PTFE-lined cap.
7.7.2 Stock solutions should be checked for signs of degradation prior to the preparation of calibration or performance test standards and shall be replaced after six months, or sooner if comparison with quality control check standards indicates a change in concentration.
7.8 Labeled compound spiking solution: From stock solutions prepared as above, or from mixtures, prepare one spiking solution to contain the labeled chlorovanillin in acetone and a second spiking solution to contain the remaining chlorophenolics, including the 3,4,5-trichlorophenol sample matrix internal standard (SMIS), in methanol. The labeled compounds and SMIS are each at a concentration of 12.5 µg/mL.
7.9 Secondary standards for calibration: Using stock solutions (Section 7.7), prepare one secondary standard containing the chlorovanillins and chlorsyringaldehydes listed in Table 1 in acetone and a second secondary standard containing the remaining chlorophenolics in methanol. The monochlorinated phenol, guaiacol, and catechol are included at a concentration of 25 µg/mL; the trichlorinated catechols, tetrachlorinated guaiacol and catechol, pentachlorophenol, 5,6-dichlorovanillin, and 2,6-dichlorosyringaldehyde are included at a concentration of 100 µg/mL; and the remaining compounds are included at a concentration of 50 µg/mL, each in their respective solutions.
7.10 Instrument internal standard (IIS): Prepare a solution of 2,2′-difluorobiphenyl (DFB) at a concentration of 2.5 mg/mL in hexane.
7.11 DFTPP solution: Prepare a solution of DFTPP at 50 µg/mL in acetone.
7.12 Solutions for obtaining authentic mass spectra (Section 10.2): Prepare mixtures of compounds at concentrations which will assure authentic spectra are obtained for storage in libraries.
7.13 Preparation of calibration solutions.
7.13.1 Into five 1000-mL aliquots of reagent water, spike 50, 100, 200, 500 and 1000 µL of each of the two solutions in Section 7.9. Spike 1.00 mL of each of the two labeled compound spiking solutions (Section 7.8) into each of the five aliquots.
7.13.2 Using the procedure in Section 11, derivatize and extract each solution, and concentrate the extract to a final volume of 0.50 mL. This will produce calibration solutions of nominal 5, 10, 20, 50, and 100 µg/mL of the native chlorophenolics and a constant concentration of 25 µg/mL of each labeled compound and the SMIS (assuming 100% derivatization and recovery). As noted in Section 11.1.6, ascorbic acid is added to all samples of final effluents to stabilize chlorocatechols, but is not added to samples of pulp and paper in-process wastewaters. Therefore, it is necessary to prepare separate sets of five initial calibration standards with and without the addition of ascorbic acid. Also, in the event that the laboratory is extracting final effluent samples by both the stir-bar and separatory funnel procedures (see Section 11.3), initial calibration standards should be prepared by both methods.
7.13.3 These solutions permit the relative response (labeled to unlabeled) and the response factor to be measured as a function of concentration (Sections 10.4 and 10.5).
7.13.4 The nominal 50 µg/mL standard may also be used as a calibration verification standard (see Section 9.6).
7.14 Ongoing precision and recovery (OPR) standard: Used for determination of initial (Section 9.3.2) and ongoing (Section 9.6) precision and recovery. This solution is prepared by spiking 500 µL of each the two solutions of the secondary calibration standards (Section 7.9) and 1 mL of each of the two labeled compound spiking solutions (Section 7.8) into 1000 mL of reagent water.
7.15 Stability of solutions: All standard solutions (Sections 7.7 through 7.14) shall be analyzed within 48 hours of preparation and on a monthly basis thereafter for signs of degradation. Standards will remain acceptable if the peak area at the quantitation m/z relative to the DFB internal standard remains within ±15% of the area obtained in the initial analysis of the standard.
8.0 Sample Collection, Preservation, and Storage8.1 Collect samples in glass containers (Section 6.1) following conventional sampling practices (Reference 9). Aqueous samples are collected in refrigerated bottles using automatic sampling equipment.
8.2 Sample preservation.
8.2.1 Residual chlorine: If the sample contains residual chlorine, the chlorine must be reduced to eliminate positive interference resulting from continued chlorination reactions. Immediately after sampling, test for residual chlorine using the following method or an alternative EPA method (Reference 10).
8.2.1.1 Dissolve a few crystals of potassium iodide in the sample and add three to five drops of a 1% starch solution. A blue color indicates the presence of residual chlorine.
8.2.1.2 If residual chlorine is found, add 1 mL of sodium thiosulfate solution (Section 7.2.1) for each 2.5 ppm of free chlorine or until the blue color disappears.
8.2.2 Acidification: Adjust pH of all aqueous samples to <2 with sulfuric acid (Section 7.1.2). Failure to acidify samples may result in positive interferences from continued chlorination reactions.
8.2.3 Refrigeration: Maintain sample temperature at 0 to 4 °C from time of collection until extraction, and maintain extracts at a temperature of 0 to 4 °C from time of extraction until analysis.
8.3 Collect a minimum of 2000 mL of sample. This will provide a sufficient amount for all testing. Smaller amounts may be collected if the stream is known to contain high levels of chlorophenolics.
8.4 All samples must be acetylated and extracted within 30 days of collection, and must be analyzed within 30 days of acetylation. If labeled compound recoveries for a sample do not meet the acceptance criteria in Table 5 and the 30-day holding time is not met, a new sample must be collected.
9.0 Quality Control9.1 Each laboratory that uses this method is required to operate a formal quality assurance program (Reference 8). The minimum requirements of this program consist of an initial demonstration of laboratory capability, analysis of samples spiked with labeled compounds to evaluate and document data quality, and analysis of standards and blanks as tests of continued performance. Laboratory performance is compared to established performance criteria to determine if the results of analyses meet the performance characteristics of the method.
9.1.1 DFTPP spectrum validity shall be checked at the beginning of each eight-hour shift during which analyses are performed. This test is described in Section 9.2.
9.1.2 The laboratory shall make an initial demonstration of the ability to generate acceptable results with this method. This ability is established as described in Section 9.3.
9.1.3 The laboratory is permitted to modify this method to improve separations or lower the costs of measurements, provided all performance specifications are met. Each time a modification is made to the method, the laboratory is required to repeat the procedures in Sections 10.3 and 9.3.2 to demonstrate method performance. If the detection limits for the analytes in this method will be affected by the modification, the laboratory should demonstrate that each MDL (40 CFR 136, appendix B) is less than or equal to the MDL in this method or one-third the regulatory compliance level, whichever is higher.
9.1.4 The laboratory shall spike all samples with labeled compounds and the sample matrix internal standard (SMIS) to monitor method performance. This test is described in Section 9.4. When results of these spikes indicate atypical method performance for samples, the samples are diluted to bring method performance within acceptable limits (Section 13).
9.1.5 Analyses of blanks are required to demonstrate freedom from contamination. The procedures and criteria for analysis of a blank are described in Section 9.5.
9.1.6 The laboratory shall, on an ongoing basis, demonstrate through analysis of the ongoing precision and recovery standard (Section 7.14) that the analysis system is in control. These procedures are described in Section 9.6.
9.1.7 The laboratory shall maintain records to define the quality of data that is generated. Development of accuracy statements is described in Section 9.4.4 and 9.6.3.
9.2 DFTPP spectrum validity: Inject 1 µL of the DFTPP solution (Section 7.11) either separately or within a few seconds of injection of the OPR standard (Section 9.6) analyzed at the beginning of each shift. The criteria in Table 3 shall be met.
9.3 Initial demonstration of laboratory capability.
9.3.1 Method Detection Limit (MDL): To establish the ability to detect the analytes in this method, the laboratory should determine the MDL per the procedure in 40 CFR 136, appendix B using the apparatus, reagents, and standards that will be used in the practice of this method. MDLs less than or equal to the MDLs in Table 2 should be achieved prior to the practice of this method.
9.3.2 Initial precision and recovery (IPR): To establish the ability to demonstrate control over the analysis system and to generate acceptable precision and accuracy, the laboratory shall perform the following operations:
9.3.2.1 Derivatize, extract, concentrate, and analyze four 1000-mL aliquots of the ongoing precision and recovery standard (OPR; Section 7.14), according to the procedure in Section 11. Separate sets of IPR aliquots must be prepared with the addition of ascorbic acid and without.
9.3.2.2 Using results of the four analyses, compute the average percent recovery (X) and the relative standard deviation of the recovery (s) for each compound, by isotope dilution for pollutants with a labeled analog, and by internal standard for pollutants with no labeled analog and for the labeled compounds and the SMIS.
9.3.2.3 For each compound, compare s and X with the corresponding limits for initial precision and recovery in Table 5. If s and X for all compounds meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may begin. If, however, any individual s exceeds the precision limit or any individual X falls outside the range for recovery, system performance is unacceptable for that compound. In this event, correct the problem and repeat the test (Section 9.3.2).
9.4 Labeled compound recovery: The laboratory shall spike all samples with labeled compounds and the sample matrix internal standard (SMIS) to assess method performance on the sample matrix.
9.4.1 Analyze each sample according to the method beginning in Section 11.
9.4.2 Compute the percent recovery (P) of the labeled compounds and the SMIS using the internal standard method (Section 14.3) with 2,2′-difluorobiphenyl as the reference compound.
9.4.3 Compare the labeled compound and SMIS recovery for each compound with the corresponding limits in Table 5. If the recovery of any compound falls outside its warning limit, method performance is unacceptable for that compound in that sample. Therefore, the sample is complex. The sample is diluted and reanalyzed per Section 13.
9.4.4 As part of the QA program for the laboratory, it is suggested, but not required, that method accuracy for samples be assessed and records maintained. After the analysis of five samples for which the labeled compounds pass the tests in Section 9.4.3, compute the average percent recovery (P) and the standard deviation of the percent recovery (sp) for the labeled compounds only. Express the accuracy assessment as a percent recovery interval from P−2sp to P = 2sp for each matrix. For example, if P = 90% and sp = 10%, the accuracy interval is expressed as 70 to 110%. Update the accuracy assessment for each compound on a regular basis (e.g., after each 20 to 30 new accuracy measurements).
9.5 Blanks: Reagent water blanks are analyzed to demonstrate freedom from contamination.
9.5.1 Extract and concentrate a 1000-mL reagent water blank with each sample batch (samples started through the extraction process on the same eight-hour shift, to a maximum of 20 samples). Blanks associated with samples to which ascorbic acid is added must be prepared with ascorbic acid, and blanks associated with samples to which ascorbic acid is not added must be prepared without ascorbic acid. Analyze the blank immediately after analysis of the OPR (Section 7.14) to demonstrate freedom from contamination.
9.5.2 If any of the compounds of interest (Table 1) or any potentially interfering compound is found in an aqueous blank at greater than 5 µg/L (assuming a response factor of one relative to the sample matrix internal standard for compounds not listed in Table 1), analysis of samples is halted until the source of contamination is eliminated and a blank shows no evidence of contamination at this level.
9.6 Calibration verification and ongoing precision and recovery: At the beginning of each eight-hour shift during which analyses are performed, analytical system performance is verified for all compounds. Analysis of DFTPP (Section 9.2) and the nominal 50 µg/mL OPR (Section 11.1.5) is used to verify all performance criteria. Adjustment and/or recalibration, per Section 10, shall be performed until all performance criteria are met. Only after all performance criteria are met may samples and blanks be analyzed.
9.6.1 Analyze the extract of the OPR (Section 11.1.5) at the beginning of each eight-hour shift and prior to analysis of samples from the same batch. Alternatively, a separate calibration verification may be performed using an aliquot of the midpoint calibration standard from Section 7.13 (with a nominal concentration of 50 µg/mL). This alternative may be used to check instrument performance on failure of an OPR, or when samples extracted with an OPR aliquot are not analyzed within the same eight-hour analysis shift.
9.6.1.1 Retention times: The absolute retention time of 2,2′-difluorobiphenyl shall be within the range of 765 to 885 seconds, and the relative retention times of all pollutants and labeled compounds shall fall within the limits given in Table 2.
9.6.1.2 GC resolution: The valley height between 4,6-dichloroguaiacol and 3,4-dichloroguaiacol at m/z 192 shall not exceed 10% of the height of the taller of the two peaks.
9.6.1.3 Multiple peaks: Each compound injected shall give a single, distinct GC peak.
9.6.2 Compute the percent recovery of each pollutant (Table 1) by isotope dilution (Section 10.4) for those compounds that have labeled analogs. Compute the percent recovery of each pollutant that has no labeled analog by the internal standard method (Section 10.5), using the 3,4,5-trichlorophenol (SMIS) as the internal standard. Compute the percent recovery of the labeled compounds and the SMIS by the internal standard method, using the 2,2′-difluorobiphenyl as the internal standard.
9.6.2.1 For each compound, compare the recovery with the limits for ongoing precision and recovery in Table 5. If all compounds meet the acceptance criteria, system performance is acceptable and analysis of blanks and samples may proceed. If, however, any individual recovery falls outside of the range given, system performance is unacceptable for that compound. In this event, there may be a problem with the GCMS or with the derivatization/extraction/concentration systems.
9.6.2.2 GCMS system: To determine if the failure of the OPR test (Section 9.6.2.1) is due to instrument drift, analyze the current calibration verification extract (Section 7.13.4), calculate the percent recoveries of all compounds, and compare with the OPR recovery limits in Table 5. If all compounds meet these criteria, GCMS performance/stability is verified, and the failure of the OPR analysis is attributed to problems in the derivatization/extraction/concentration of the OPR. In this case, analysis of the sample extracts may proceed. However, failure of any of the recovery criteria in the analysis of a sample extract requires rederivatization of that sample (Sections 13.3.1 and 13.3.2). If, however, the performance/stability of the GCMS is not verified by analysis of the calibration verification extract, the GCMS requires recalibration and all extracts associated with the failed OPR must be reanalyzed.
9.6.3 Add results that pass the specifications in Section 9.6.2.1 to initial and previous ongoing data for each compound. Update QC charts to form a graphic representation of continued laboratory performance. Develop a statement of laboratory accuracy for each pollutant and labeled compound in each matrix type (reagent water, C-stage filtrate, E-stage filtrate, final effluent, etc.) by calculating the average percent recovery (R) and the standard deviation of percent recovery (sr). Express the accuracy as a recovery interval from R− 2sr to R = 2sr. For example, if R = 95% and sr = 5%, the accuracy is 85 to 105%.
9.7 The specifications contained in this method can be met if the apparatus used is calibrated properly, then maintained in a calibrated state. The standards used for calibration (Section 10) and for initial (Section 9.3.2) and ongoing (Section 9.6) precision and recovery should be identical, so that the most precise results will be obtained. The GCMS instrument in particular will provide the most reproducible results if dedicated to the settings and conditions required for the analyses of chlorophenolics by this method.
9.8 Depending on specific program requirements, field replicates may be collected to determine the precision of the sampling technique, and spiked samples may be required to determine the accuracy of the analysis when the internal standard method is used.
10.0 Calibration and Standardization10.1 Assemble the GCMS and establish the operating conditions in Section 12. Analyze standards per the procedure in Section 12 to demonstrate that the analytical system meets the minimum levels in Table 2, and the mass-intensity criteria in Table 3 for 50 ng DFTPP.
10.2 Mass-spectral libraries: Detection and identification of compounds of interest are dependent upon spectra stored in user-created libraries.
10.2.1 Obtain a mass spectrum of the acetyl derivative of each chlorophenolic compound (pollutant, labeled compound, and the sample matrix internal standard) by derivatizing and analyzing an authentic standard either singly or as part of a mixture in which there is no interference between closely eluting components. That only a single compound is present is determined by examination of the spectrum. Fragments not attributable to the compound under study indicate the presence of an interfering compound.
10.2.2 Adjust the analytical conditions and scan rate (for this test only) to produce an undistorted spectrum at the GC peak maximum. An undistorted spectrum will usually be obtained if five complete spectra are collected across the upper half of the GC peak. Software algorithms designed to “enhance” the spectrum may eliminate distortion, but may also eliminate authentic m/z's or introduce other distortion.
10.2.3 The authentic reference spectrum is obtained under DFTPP tuning conditions (Section 10.1 and Table 3) to normalize it to spectra from other instruments.
10.2.4 The spectrum is edited by removing all peaks in the m/z 42 to 45 range, and saving the five most intense mass spectral peaks and all other mass spectral peaks greater than 10% of the base peak (excluding the peaks in the m/z 42 to 45 range). The spectrum may be further edited to remove common interfering m/z's. The spectrum obtained is stored for reverse search and for compound confirmation. 10.3 Minimum level: Demonstrate that the chlorophenolics are detectable at the minimum level (per all criteria in Section 14). The nominal 5 µg/mL calibration standard (Section 7.13) can be used to demonstrate this performance.
10.4 Calibration with isotope dilution: Isotope dilution is used when (1) labeled compounds are available, (2) interferences do not preclude its use, and (3) the quantitation m/z (Table 4) extracted ion-current profile (EICP) area for the compound is in the calibration range. Alternative labeled compounds and quantitation m/z's may be used based on availability. If any of the above conditions preclude isotope dilution, the internal standard calibration method (Section 10.5) is used.
10.4.1 A calibration curve encompassing the concentration range is prepared for each compound to be determined. The relative response (pollutant to labeled) vs. concentration in standard solutions is plotted or computed using a linear regression. The example in Figure 1 shows a calibration curve for phenol using phenol-d5 as the isotopic diluent. Also shown are the ±10% error limits (dotted lines). Relative response (RR) is determined according to the procedures described below. A minimum of five data points are employed for calibration.
10.4.2 The relative response of a pollutant to its labeled analog is determined from isotope ratio values computed from acquired data. Three isotope ratios are used in this process:
Rx = the isotope ratio measured for the pure pollutant. Ry = the isotope ratio measured for the labeled compound. Rm = the isotope ratio of an analytical mixture of pollutant and labeled compounds.The m/z's are selected such that Rx>Ry. If Rm is not between 2Ry and 0.5Rx, the method does not apply and the sample is analyzed by the internal standard method.
10.4.3 Capillary columns sometimes separate the pollutant-labeled pair when deuterium labeled compounds are used, with the labeled compound eluted first (Figure 2). For this case,

10.4.4 When the pollutant-labeled pair is not separated (as occurs with carbon-13-labeled compounds), or when another labeled compound with interfering spectral masses overlaps the pollutant (a case which can occur with isomeric compounds), it is necessary to determine the contributions of the pollutant and labeled compound to the respective EICP areas. If the peaks are separated well enough to permit the data system or operator to remove the contributions of the compounds to each other, the equations in Section 10.4.3 apply. This usually occurs when the height of the valley between the two GC peaks at the same m/z is less than 70 to 90% of the height of the shorter of the two peaks. If significant GC and spectral overlap occur, RR is computed using the following equation:
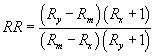 Where:
Rx is measured as shown in figure 3A, Ry is measured
as shown in figure 3B, Rm is measured as shown in figure 3C.
Where:
Rx is measured as shown in figure 3A, Ry is measured
as shown in figure 3B, Rm is measured as shown in figure 3C.
For example, Rx = 46100/4780 = 9.644; Ry = 2650/43600 = 0.0608; Rm = 49200/48300 = 1.1019; thus, RR = 1.114. 10.4.5 To calibrate the analytical system by isotope dilution, analyze a 1-µL aliquot of each of the calibration standards (Section 7.13) using the procedure in Section 12. Compute the RR at each concentration.
10.4.6 Linearity: If the ratio of relative response to concentration for any compound is constant (less than 20% coefficient of variation) over the five-point calibration range, an averaged relative response/concentration ratio may be used for that compound; otherwise, the complete calibration curve for that compound shall be used over the five-point calibration range.
10.5 Calibration by internal standard: The method contains two types of internal standards, the sample matrix internal standard (SMIS) and the instrument internal standard (IIS), and they are used for different quantitative purposes. The 3,4,5-trichlorophenol sample matrix internal standard (SMIS) is used for measurement of all pollutants with no labeled analog and when the criteria for isotope dilution (Section 10.4) cannot be met. The 2,2′-difluorobiphenyl instrument internal standard (IIS) is used for determination of the labeled compounds and the SMIS. The results are used for intralaboratory statistics (Sections 9.4.4 and 9.6.3).
10.5.1 Response factors: Calibration requires the determination of response factors (RF) for both the pollutants with no labeled analog and for the labeled compounds and the SMIS. The response factor is defined by the following equation:
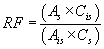 Where:
As = the area of the characteristic mass for the compound in
the daily standard. Ais = the area of the characteristic
mass for the internal standard. Cis = the concentration of
the internal standard (µg/mL). Cs = is the concentration of
the compound in the calibration standard (µg/mL).
Where:
As = the area of the characteristic mass for the compound in
the daily standard. Ais = the area of the characteristic
mass for the internal standard. Cis = the concentration of
the internal standard (µg/mL). Cs = is the concentration of
the compound in the calibration standard (µg/mL).
When this equation is used to determine the response factors for pollutant compounds without labeled analogs, use the area (Ais) and concentration (Cis) of 3,4,5-trichlorophenol (SMIS) as the internal standard. When this equation is used to determine the response factors for the labeled analogs and the SMIS, use the area (Ais) and concentration (Cis) of 2,2′-difluorobiphenyl as the internal standard.
10.5.2 The response factor is determined for at least five concentrations appropriate to the response of each compound (Section 7.13); nominally, 5, 10, 20, 50, and 100 µg/mL. The amount of SMIS added to each solution is the same (25 µg/mL) so that Cis remains constant. Likewise, the concentration of IIS is constant in each solution. The area ratio (As/Ais) is plotted versus the concentration ratio (Cs/Cis) for each compound in the standard to produce a calibration curve.
10.5.3 Linearity: If the response factor (RF) for any compound is constant (less than 35% coefficient of variation) over the five-point calibration range, an averaged response factor may be used for that compound; otherwise, the complete calibration curve for that compound shall be used over the five-point range.
10.6 Combined calibration: By using calibration solutions (Section 7.13) containing the pollutants, labeled compounds, and the internal standards, a single set of analyses can be used to produce calibration curves for the isotope dilution and internal standard methods. These curves are verified each shift (Section 9) by analyzing the OPR standard, or an optional calibration verification (VER) standard. Recalibration is required only if OPR criteria (Section 9.6 and Table 5) cannot be met.
11.0 Sample Derivatization, Extraction, and ConcentrationThe procedure described in this section uses a stir-bar in a beaker for the derivatization. The extraction procedures applied to samples depend on the type of sample being analyzed. Extraction of samples from in-process wastewaters is performed using a separatory funnel procedure. All calibrations, IPR, OPR, and blank analyses associated with in-process wastewater samples must be performed by the separatory funnel procedure.
Extraction of samples of final effluents and raw water may be performed using either the stir-bar procedure or the separatory funnel procedure. However, all calibrations, IPR, OPR, blank, and sample analyses must be performed using the same procedure. Both procedures are described below.
11.1 Preparation of all sample types for stir-bar derivatization.
11.1.1 Allow sample to warm to room temperature.
11.1.2 Immediately prior to measuring, shake sample vigorously to insure homogeneity.
11.1.3 Measure 1000 mL (±10 mL) of sample into a clean 2000-mL beaker. Label the beaker with the sample number.
11.1.4 Dilute aliquot(s).
11.1.4.1 Complex samples: For samples that are expected to be difficult to derivatize, concentrate, or are expected to overload the GC column or mass spectrometer, measure an additional 100 mL (±1 mL) into a clean 2000-mL beaker and dilute to a final volume of 1000-mL (±50 mL) with reagent water. Label with the sample number and as the dilute aliquot. However, to ensure adequate sensitivity, a 1000-mL aliquot must always be prepared and analyzed.
11.1.4.2 Pulp and paper industry samples: For in-process streams such as E-stage and C-stage filtrates and other in-process wastewaters, it may be necessary to prepare an aliquot at an additional level of dilution. In this case, dilute 10 mL (±0.1 mL) of sample to 1000-mL (±50 mL).
11.1.5 QC aliquots: For a batch of samples of the same type to be extracted at the same time (to a maximum of 20), place two 1000-mL (±10 mL) aliquots of reagent water in clean 2000-mL beakers. Label one beaker as the blank and the other as the ongoing precision and recovery (OPR) aliquot. Because final effluent samples are treated with ascorbic acid and in-process wastewater samples are not (see Section 11.1.6), prepare an OPR aliquot and a blank for the final effluent and a separate pair for the in-process samples. Treat these QC aliquots in the same fashion as the associated samples, adding ascorbic acid to the pair associated with the final effluents, and not adding ascorbic acid to the pair associated with the in-process samples.
11.1.6 Ascorbic acid: Added to stabilize chlorocatechols. However, for pulp and paper industry in-process streams and other in-process wastewaters, the addition of ascorbic acid may convert chloro-o-quinones to catechols if these quinones are present. Separate calibration curves must be prepared with and without the addition of ascorbic acid (Section 7.13.2).
11.1.6.1 Spike 5 to 6 mL of the ascorbic acid solution (Section 7.2.2) into each final effluent sample, and the associated calibration standards, IPR and OPR aliquots, and blank.
11.1.6.2 For pulp and paper industry C-stage filtrates, E-stage filtrates, and untreated effluents, omit the ascorbic acid to prevent the conversion of chloro-o-quinones to catechols. Prepare calibration standards, IPR and OPR aliquots, and blanks associated with these samples without ascorbic acid as well.
11.1.7 Spike 1000 µL of the labeled compound spiking solution (Section 7.8) into the sample and QC aliquots.
11.1.8 Spike 500 µL of the nominal 50 µg/mL calibration solution (Section 7.13.4) into the OPR aliquot.
11.1.9 Adjust the pH of the sample aliquots to between 7.0 and 7.1. For calibration standards, IPR and OPR aliquots, and blanks, pH adjustment is not required.
11.1.10 Equilibrate all sample and QC solutions for approximately 15 minutes, with occasional stirring.
11.2 Derivatization: Because derivatization must proceed rapidly, particularly upon the addition of the K2CO3 buffer, it is necessary to work with one sample at a time until the derivatization step (Section 11.2.3) is complete.
11.2.1 Place a beaker containing a sample or QC aliquot on the magnetic stirrer in a fume hood, drop a clean stirring bar into the beaker, and increase the speed of the stirring bar until the vortex is drawn to the bottom of the beaker.
11.2.2 Measure 25 to 26 mL of K2CO3 buffer into a graduated cylinder or other container and 25 to 26 mL of acetic acid into another.
11.2.3 Add the K2CO3 buffer to the sample or QC aliquot, immediately (within one to three seconds) add the acetic anhydride, and stir for three to five minutes to complete the derivatization.
11.3 Extraction: Two procedures are described below for the extraction of derivatized samples. The choice of extraction procedure will depend on the sample type. For final effluent samples, either of two procedures may be utilized for extraction of derivatized samples. For samples of in-process wastewaters, the separatory funnel extraction procedure must be used.
Note:Whichever procedure is employed, the same extraction procedure must be used for calibration standards, IPR aliquots, OPR aliquots, blanks, and the associated field samples.
11.3.1 Stir-bar extraction of final effluents.
11.3.1.1 Add 200 mL (±20 mL) of hexane to the beaker and stir for three to five minutes, drawing the vortex to the bottom of the beaker.
11.3.1.2 Stop the stirring and drain the hexane and a portion of the water into a 500-to 1000-mL separatory funnel. Allow the layers to separate.
11.3.1.3 Drain the aqueous layer back into the beaker.
11.3.1.4 The formation of emulsions can be expected in any solvent extraction procedure. If an emulsion forms, the laboratory must take steps to break the emulsion before proceeding. Mechanical means of breaking the emulsion include the use of a glass stirring rod, filtration through glass wool, and other techniques. For emulsions that resist these techniques, centrifugation is nearly 100% effective.
If centrifugation is employed to break the emulsion, drain the organic layer into a centrifuge tube, cap the tube, and centrifuge for two to three minutes or until the phases separate. If the emulsion cannot be completely broken, collect as much of the organic phase as possible, and measure and record the volume of the organic phase collected.
If all efforts to break the emulsion fail, including centrifugation, and none of the organic phase can be collected, proceed with the dilute aliquot (Section 11.1.4.2). However, use of the dilute aliquot will sacrifice the sensitivity of the method, and may not be appropriate in all cases.
11.3.1.5 Drain the organic layer into a Kuderna-Danish (K-D) apparatus equipped with a 10-mL concentrator tube. Label the K-D apparatus. It may be necessary to pour the organic layer through a funnel containing anhydrous sodium sulfate to remove any traces of water from the extract.
11.3.1.6 Repeat the extraction (Section 11.3.1.1 through 11.3.1.5) two more times using another 200-mL of hexane for each extraction, combining the extracts in the K-D apparatus.
11.3.1.7 Proceed with concentration of the extract, as described in Section 11.4.
11.3.2 Separatory funnel extraction of either final effluents or in-process wastewaters.
11.3.2.1 Transfer the derivatized sample or QC aliquot to a 2-L separatory funnel.
11.3.2.2 Add 200 mL (±20 mL) of hexane to the separatory funnel. Cap the funnel and extract the sample by shaking the funnel for two to three minutes with periodic venting.
11.3.2.3 Allow the organic layer to separate from the water phase for a minimum of 10 minutes.
11.3.2.4 Drain the lower aqueous layer into the beaker used for derivatization (Section 11.2), or into a second clean 2-L separatory funnel. Transfer the solvent to a 1000-mL K-D flask. It may be necessary to pour the organic layer through a funnel containing anhydrous sodium sulfate to remove any traces of water from the extract.
11.3.2.5 The formation of emulsions can be expected in any solvent extraction procedure. If an emulsion forms, the laboratory must take steps to break the emulsion before proceeding. Mechanical means of breaking the emulsion include the use of a glass stirring rod, filtration through glass wool, and other techniques. For emulsions that resist these techniques, centrifugation may be required.
If centrifugation is employed to break the emulsion, drain the organic layer into a centrifuge tube, cap the tube, and centrifuge for two to three minutes or until the phases separate. If the emulsion cannot be completely broken, collect as much of the organic phase as possible, and measure and record the volume of the organic phase collected. If all efforts to break the emulsion, including centrifugation, fail and none of the organic phase can be collected, proceed with the dilute aliquot (Section 11.1.4.2). However, use of the dilute aliquot will sacrifice the sensitivity of the method, and may not be appropriate in all cases.
11.3.2.6 If drained into a beaker, transfer the aqueous layer to the 2-L separatory funnel (Section 11.3.2.1). Perform a second extraction using another 200 mL of fresh solvent.
11.3.2.7 Transfer the extract to the 1000-mL K-D flask in Section 11.3.2.4.
11.3.2.8 Perform a third extraction in the same fashion as above.
11.3.2.9 Proceed with concentration of the extract, as described in Section 11.4.
11.4 Macro concentration: Concentrate the extracts in separate 1000-mL K-D flasks equipped with 10-mL concentrator tubes. Add one to two clean boiling chips to the flask and attach a three-ball macro-Snyder column. Prewet the column by adding approximately 1 mL of hexane through the top. Place the K-D apparatus in a hot water bath so that the entire lower rounded surface of the flask is bathed with steam. Adjust the vertical position of the apparatus and the water temperature as required to complete the concentration in 15 to 20 minutes. At the proper rate of distillation, the balls of the column will actively chatter but the chambers will not flood. When the liquid has reached an apparent volume of 1 mL, remove the K-D apparatus from the bath and allow the solvent to drain and cool for at least 10 minutes. Remove the Snyder column and rinse the flask and its lower joint into the concentrator tube with 1 to 2 mL of hexane. A 5-mL syringe is recommended for this operation.
11.5 Micro-concentration: Final concentration of the extracts may be accomplished using either a micro-Snyder column or nitrogen evaporation.
11.5.1 Micro-Snyder column: Add a clean boiling chip and attach a two-ball micro-Snyder column to the concentrator tube. Prewet the column by adding approximately 0.5 mL hexane through the top. Place the apparatus in the hot water bath. Adjust the vertical position and the water temperature as required to complete the concentration in 5 to 10 minutes. At the proper rate of distillation, the balls of the column will actively chatter but the chambers will not flood. When the liquid reaches an apparent volume of approximately 0.2 mL, remove the apparatus from the water bath and allow to drain and cool for at least 10 minutes. Remove the micro-Snyder column and rinse its lower joint into the concentrator tube with approximately 0.2 mL of hexane. Adjust to a final volume of 0.5 mL.
11.5.2 Nitrogen evaporation: Transfer the concentrator tube to a nitrogen evaporation device and direct a gentle stream of clean dry nitrogen into the concentrator. Rinse the sides of the concentrator tube with small volumes of hexane, and concentrate the extract to a final volume of 0.5 mL.
11.6 Spike each extract with 10 µL of the 2,2′-difluorobiphenyl IIS (Section 7.10) and transfer the concentrated extract to a clean screw-cap vial using hexane to rinse the concentrator tube. Seal the vial with a PTFE-lined lid, and mark the level on the vial. Label with the sample number and store in the dark at −20 to −10 °C until ready for analysis.
12.0 GCMS Analysis12.1 Establish the following operating conditions:
Carrier gas flow: Helium at 30 cm/sec at 50 °C Injector temperature: 300 °C Initial temperature: 50 °C Temperature program: 8 °C/min to 270 °C Final hold: Until after 2,6-dichlorosyringaldehyde elutesAdjust the GC conditions to meet the requirements in Section 9.6.1.1 and Table 2 for analyte separation and sensitivity. Once optimized, the same GC conditions must be used for the analysis of all standards, blanks, IPR and OPR aliquots, and samples.
12.2 Bring the concentrated extract (Section 11.6) or standard (Sections 7.13 and 7.14) to room temperature and verify that any precipitate has redissolved. Verify the level on the extract (Sections 7.13, 7.14, and 11.6) and bring to the mark with solvent if required.
12.3 Inject a 1-µL volume of the standard solution or extract using on-column or splitless injection. For 0.5 mL extracts, this 1-µL injection volume will contain 50 ng of the DFB internal standard. If an injection volume other than 1 µL is used, that volume must contain 50 ng of DFB.
12.4 Start the GC column temperature ramp upon injection. Start MS data collection after the solvent peak elutes. Stop data collection after the 2,6-dichlorosyringaldehyde peak elutes. Return the column to the initial temperature for analysis of the next sample.
13.0 Analysis of Complex SamplesSome samples may contain high levels (>1000 µg/L) of the compounds of interest, interfering compounds, and/or other phenolic materials. Some samples will not concentrate to 0.5 mL (Section 11.5); others will overload the GC column and/or mass spectrometer; others may contain amounts of phenols that may exceed the capacity of the derivatizing agent.
13.1 Analyze the dilute aliquot (Section 11.1.4) when the sample will not concentrate to 0.5 mL. If a dilute aliquot was not extracted, and the sample holding time (Section 8.4) has not been exceeded, dilute an aliquot of sample with reagent water, and derivatize and extract it (Section 11.1.4). Otherwise, dilute the extract (Section 14.7.3) and quantitate it by the internal standard method (Section 14.3).
13.2 Recovery of the 2,2′-difluorobiphenyl instrument internal standard: The EICP area of the internal standard should be within a factor of two of the area in the OPR or VER standard (Section 9.6). If the absolute areas of the labeled compounds and the SMIS are within a factor of two of the respective areas in the OPR or VER standard, and the DFB internal standard area is less than one-half of its respective area, then internal standard loss in the extract has occurred. In this case, analyze the extract from the dilute aliquot (Section 11.1.4).
13.3 Recovery of labeled compounds and the sample matrix internal standard (SMIS): SMIS and labeled compound recovery specifications have been developed for samples with and without the addition of ascorbic acid. Compare the recoveries to the appropriate limits in Table 5.
13.3.1 If SMIS or labeled compound recoveries are outside the limits given in Table 5 and the associated OPR analysis meets the recovery criteria, the extract from the dilute aliquot (Section 11.1.4) is analyzed as in Section 14.7.
13.3.2 If labeled compound or SMIS recovery is outside the limits given in Table 5 and the associated OPR analysis did not meet recovery criteria, a problem in the derivatization/extraction/concentration of the sample is indicated, and the sample must be rederivatized and reanalyzed.
14.0 Data Analysis and Calculations14.1 Qualitative determination: Identification is accomplished by comparison of data from analysis of a sample or blank with data stored in the mass spectral libraries. Identification of a compound is confirmed when the following criteria are met:
14.1.1 The signals for m/z 43 (to indicate the presence of the acetyl derivative) and all characteristic m/z's stored in the spectral library (Section 10.2.4) shall be present and shall maximize within the same two consecutive scans.
14.1.2 Either (1) the background corrected EICP areas, or (2) the corrected relative intensities of the mass spectral peaks at the GC peak maximum shall agree within a factor of two (0.5 to 2 times) for all m/z's stored in the library.
14.1.3 The relative retention time shall be within the window specified in Table 2.
14.1.4 The m/z's present in the mass spectrum from the component in the sample that are not present in the reference mass spectrum shall be accounted for by contaminant or background ions. If the mass spectrum is contaminated, an experienced spectrometrist (Section 1.4) shall determine the presence or absence of the compound.
14.2 Quantitative determination by isotope dilution: By adding a known amount of a labeled compound to every sample prior to derivatization and extraction, correction for recovery of the pollutant can be made because the pollutant and its labeled analog exhibit the same effects upon derivatization, extraction, concentration, and gas chromatography. Relative response (RR) values for sample mixtures are used in conjunction with calibration curves described in Section 10.4 to determine concentrations directly, so long as labeled compound spiking levels are constant. For the phenol example given in Figure 1 (Section 10.4.1), RR would be equal to 1.114. For this RR value, the phenol calibration curve given in Figure 1 indicates a concentration of 27 µg/mL in the sample extract (Cex).
14.2.1 Compute the concentration in the extract using the response ratio determined from calibration data (Section 10.4) and the following equation:
 Where:
Cex = concentration of the pollutant in the extract. An = area of
the characteristic m/z for the pollutant. Cl = concentration of the
labeled compound in the extract. Al = area of the characteristic
m/z for the labeled compound. RR = response ratio from the initial
calibration.
Where:
Cex = concentration of the pollutant in the extract. An = area of
the characteristic m/z for the pollutant. Cl = concentration of the
labeled compound in the extract. Al = area of the characteristic
m/z for the labeled compound. RR = response ratio from the initial
calibration.
14.2.2 For the IPR (Section 9.3.2) and OPR (Section 9.6), compute the percent recovery of each pollutant using the equation in Section 14.6. The percent recovery is used for the evaluation of method and laboratory performance, in the form of IPR (Section 9.3.2) and OPR (Section 9.6).
14.3 Quantitative determination by internal standard: Compute the concentration using the response factor determined from calibration data (Section 10.5) and the following equation:
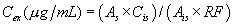 Where:
Cex = concentration of the pollutant in the extract. As = area of
the characteristic m/z for the pollutant. Cis = concentration of
the internal standard in the extract (see note below). Ais = area
of the characteristic m/z for the internal standard. RF = response
factor from the initial calibration. Note:
Where:
Cex = concentration of the pollutant in the extract. As = area of
the characteristic m/z for the pollutant. Cis = concentration of
the internal standard in the extract (see note below). Ais = area
of the characteristic m/z for the internal standard. RF = response
factor from the initial calibration. Note:
When this equation is used to compute the extract concentrations of native compounds without labeled analogs, use the area (Ais) and concentration (Cis) of 3,4,5-trichlorophenol (SMIS) as the internal standard.
For the IPR (Section 9.3.2) and OPR (Section 9.6), compute the percent recovery using the equation in Section 14.6.
Note:Separate calibration curves will be required for samples with and without the addition of ascorbic acid, and also for both extraction procedures (stir-bar and separatory funnel) where applicable.
14.4 Compute the concentration of the labeled compounds and the SMIS using the equation in Section 14.3, but using the area and concentration of the 2,2′-difluorobiphenyl as the internal standard, and the area of the labeled compound or SMIS as As.
14.5 Compute the concentration of each pollutant compound in the sample using the following equation:
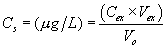 Where: Cs
= Concentration of the pollutant in the sample. Cex = Concentration
of the pollutant in the extract. Vex = Volume of the concentrated
extract (typically 0.5 mL). Vo = Volume of the original sample in
liters.
Where: Cs
= Concentration of the pollutant in the sample. Cex = Concentration
of the pollutant in the extract. Vex = Volume of the concentrated
extract (typically 0.5 mL). Vo = Volume of the original sample in
liters.
14.6 Compute the recovery of each labeled compound and the SMIS as the ratio of concentration (or amount) found to the concentration (or amount) spiked, using the following equation:

These percent recoveries are used to assess method performance according to Sections 9 and 13.
14.7 If the EICP area at the quantitation m/z for any compound exceeds the calibration range of the system, three approaches are used to obtain results within the calibration range.
14.7.1 If the recoveries of all the labeled compounds in the original sample aliquot meet the limits in Table 5, then the extract of the sample may be diluted by a maximum of a factor of 10, and the diluted extract reanalyzed.
14.7.2 If the recovery of any labeled compound is outside its limits in Table 5, or if a tenfold dilution of the extract will not bring the pollutant within the calibration range, then extract and analyze a dilute aliquot of the sample (Section 11). Dilute 100 mL, 10 mL, or an appropriate volume of sample to 1000 mL with reagent water and extract per Section 11.
14.7.3 If the recoveries of all labeled compounds in the original sample aliquot (Section 14.7.1) meet the limits in Table 5, and if the sample holding time has been exceeded, then the original sample extract is diluted by successive factors of 10, the DFB internal standard is added to give a concentration of 50 µg/mL in the diluted extract, and the diluted extract is analyzed. Quantitation of all analytes is performed using the DFB internal standard.
14.7.4 If the recoveries of all labeled compounds in the original sample aliquot (Section 14.7.1) or in the dilute aliquot (Section 14.7.2) (if a dilute aliquot was analyzed) do not meet the limits in Table 5, and if the holding time has been exceeded, re-sampling is required.
14.8 Results are reported for all pollutants, labeled compounds, and the sample matrix internal standard in standards, blanks, and samples, in units of µg/L.
14.8.1 Results for samples which have been diluted are reported at the least dilute level at which the area at the quantitation m/z is within the calibration range (Section 14.7).
14.8.2 For compounds having a labeled analog, results are reported at the least dilute level at which the area at the quantitation m/z is within the calibration range (Section 14.7) and the labeled compound recovery is within the normal range for the method (Section 13.3).
15.0 Method Performance15.1 Single laboratory performance for this method is detailed in References 1, 2, and 11. Acceptance criteria were established from multiple laboratory use of the draft method.
15.2 A chromatogram of the ongoing precision and recovery standard (Section 7.14) is shown in Figure 4.
16.0 Pollution Prevention16.1 The solvents used in this method pose little threat to the environment when recycled and managed properly.
16.2 Standards should be prepared in volumes consistent with laboratory use to minimize the volume of expired standards to be disposed.
17.0 Waste Management17.1 It is the laboratory's responsibility to comply with all federal, state, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions, and to protect the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. Compliance with all sewage discharge permits and regulations is also required.
17.2 Samples preserved with HCl or H2SO4 to pH <2 are hazardous and must be neutralized before being disposed, or must be handled as hazardous waste.
17.3 For further information on waste management, consult “The Waste Management Manual for Laboratory Personnel”, and “Less is Better: Laboratory Chemical Management for Waste Reduction”, both available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street N.W., Washington, DC 20036.
18.0 References18.1 “Chlorinated Phenolics in Water by In Situ Acetylation/GC/MS Determination,” Method CP-86.01, National Council of the Paper Industry for Air and Stream Improvement, Inc., 260 Madison Avenue, New York, NY 10016 (July 1986).
18.2 “6240-Chlorinated Phenolics (Interim Standard),” Draft Version, U.S. Environmental Protection Agency, Manchester Laboratory, Manchester, Washington.
18.3 “Performance Tests for the Evaluation of Computerized Gas Chromatography/Mass Spectrometry Equipment and Laboratories,” USEPA, EMSL Cincinnati, OH 45268, EPA-600/4-80-025 (April 1980).
18.4 “Working with Carcinogens,” DHEW, PHS, CDC, NIOSH, Publication 77-206 (August 1977).
18.5 “OSHA Safety and Health Standards, General Industry,” OSHA 2206, 29 CFR 1910 (January 1976).
18.6 “Safety in Academic Chemistry Laboratories,” ACS Committee on Chemical Safety (1979).
18.7 “Interlaboratory Validation of U. S. Environmental Protection Agency Method 1625A, Addendum Report,” SRI International, Prepared for Analysis and Evaluation Division (WH-557), USEPA, 401 M St., SW., Washington, DC 20460 (January 1985).
18.8 “Handbook of Analytical Quality Control in Water and Wastewater Laboratories,” USEPA, EMSL, Cincinnati, OH 45268, EPA-600/4-79-019 (March 1979).
18.9 “Standard Practice for Sampling Water,” ASTM Annual Book of Standards, ASTM, Philadelphia, PA, 76 (1980).
18.10 “Methods 330.4 and 330.5 for Total Residual Chlorine,” USEPA, EMSL, Cincinnati, OH 45268, EPA 600/4-70-020 (March 1979).
18.11 “Determination of Chlorophenolics, Special Analytical Services Contract 1047, Episode 1886,” Analytical Technologies, Inc., Prepared for W. A. Telliard, Industrial Technology Division (WH-552), USEPA, 401 M St., SW., Washington, DC 20460 (June 1990).
18.12 “Determination of Chlorophenolics by GCMS, Development of Method 1653,” Analytical Technologies, Inc., Prepared for W. A. Telliard, Industrial Technology Division (WH-552), USEPA, 401 M St., SW., Washington, DC 20460 (May 1991).
19.0 Tables and FiguresTable 1 - Chlorophenolic Compounds Determined by GCMS Using Isotope Dilution and Internal Standard Techniques
| Compound | Pollutant | Labeled compound | |||
|---|---|---|---|---|---|
| CAS registry | EPA-EGD | Analog | CAS registry | EPA-EGD | |
| 4-chlorophenol | 106-48-9 | 1001 | |||
| 2,4-dichlorophenol | 120-83-2 | 1002 | d3 | 93951-74-7 | 1102 |
| 2,6-dichlorophenol | 87-65-0 | 1003 | |||
| 2,4,5-trichlorophenol | 95-95-4 | 1004 | |||
| 2,4,6-trichlorophenol | 88-06-2 | 1005 | |||
| 2,3,4,6-tetrachlorophenol | 58-90-2 | 1006 | |||
| pentachlorophenol | 87-86-5 | 1007 | 13C6 | 85380-74-1 | 1107 |
| 4-chloroguaiacol | 16766-30-6 | 1008 | 13C6 | 136955-39-0 | 1108 |
| 3,4-dichloroguaiacol | 77102-94-4 | 1009 | |||
| 4,5-dichloroguaiacol | 2460-49-3 | 1010 | |||
| 4,6-dichloroguaiacol | 16766-31-7 | 1011 | |||
| 3,4,5-trichloroguaiacol | 57057-83-7 | 1012 | |||
| 3,4,6-trichloroguaiacol | 60712-44-9 | 1013 | |||
| 4,5,6-trichloroguaiacol | 2668-24-8 | 1014 | 13C6 | 136955-40-3 | 1114 |
| tetrachloroguaiacol | 2539-17-5 | 1015 | 13C6 | 136955-41-4 | 1115 |
| 4-chlorocatechol | 2138-22-9 | 1016 | |||
| 3,4-dichlorocatechol | 3978-67-4 | 1017 | |||
| 3,6-dichlorocatechol | 3938-16-7 | 1018 | |||
| 4,5-dichlorocatechol | 3428-24-8 | 1019 | 13C6 | 136955-42-5 | 1119 |
| 3,4,5-trichlorocatechol | 56961-20-7 | 1020 | |||
| 3,4,6-trichlorocatechol | 32139-72-3 | 1021 | |||
| tetrachlorocatechol | 1198-55-6 | 1022 | 13C6 | 136955-43-6 | 1122 |
| 5-chlorovanillin | 19463-48-0 | 1023 | 13C6 | 136955-44-7 | 1123 |
| 6-chlorovanillin | 18268-76-3 | 1024 | |||
| 5,6-dichlorovanillin | 18268-69-4 | 1025 | |||
| 2-chlorosyringaldehyde | 76341-69-0 | 1026 | |||
| 2,6-dichlorosyringaldehyde | 76330-06-8 | 1027 | |||
| trichlorosyringol | 2539-26-6 | 1028 | |||
| Sample matrix internal standard (SMIS) | |||||
| 3,4,5-trichlorophenol | 609-19-8 | 184 | |||
| Instrument internal standard (IIS) | |||||
| 2,2′-difluorobiphenyl | 388-82-9 | 164 | |||
Table 2 - Gas Chromatography and Method Detection Limits for Chlorophenolics
| EGD No. 1 | Compound | Retention time mean (sec) 2 | EGD ref No. |
RRT window 3 |
Minimum level 4 (µg/L) | MDL 5 (µg/L) |
|---|---|---|---|---|---|---|
| 1001 | 4-chlorophenol | 691 | 184 | 0.651-0.681 | 1.25 | 1.11 |
| 1003 | 2,6-dichlorophenol | 796 | 184 | 0.757-0.779 | 2.5 | 1.39 |
| 1102 | 2,4-dichlorophenol-d3 | 818 | 164 | 0.986-0.998 | ||
| 1202 | 2,4-dichlorophenol | 819 | 1102 | 0.997-1.006 | 2.5 | 0.15 |
| 164 | 2,2′-difluorobiphenyl (I.S.) | 825 | 164 | 1.000 | ||
| 1108 | 4-chloroguaiacol- 13C6 | 900 | 164 | 1.077-1.103 | ||
| 1208 | 4-chloroguaiacol | 900 | 1108 | 0.998-1.002 | 1.25 | 0.09 |
| 1005 | 2,4,6-trichlorophenol | 920 | 184 | 0.879-0.895 | 2.5 | 0.71 |
| 1004 | 2,4,5-trichlorophenol | 979 | 184 | 0.936-0.952 | 2.5 | 0.57 |
| 1016 | 4-chlorocatechol | 1004 | 184 | 0.961-0.975 | 1.25 | 0.59 |
| 1011 | 4,6-dichloroguaiacol | 1021 | 184 | 0.979-0.991 | 2.5 | 0.45 |
| 1009 | 3,4-dichloroguaiacol | 1029 | 184 | 0.986-0.998 | 2.5 | 0.52 |
| 184 | 3,4,5-trichlorophenol (I.S.) | 1037 | 164 | 1.242-1.272 | ||
| 1010 | 4,5-dichloroguaiacol | 1071 | 184 | 1.026-1.040 | 2.5 | 0.52 |
| 1018 | 3,6-dichlorocatechol | 1084 | 184 | 1.037-1.053 | 2.5 | 0.57 |
| 1006 | 2,3,4,6-tetrachlorophenol | 1103 | 184 | 1.050-1.078 | 2.5 | 0.38 |
| 1123 | 5-chlorovanillin- 13C6 | 1111 | 164 | 1.327-1.367 | ||
| 1223 | 5-chlorovanillin | 1111 | 1123 | 0.998-1.001 | 2.5 | 1.01 |
| 1013 | 3,4,6-trichloroguaiacol | 1118 | 184 | 1.066-1.090 | 2.5 | 0.46 |
| 1024 | 6-chlorovanillin | 1122 | 184 | 1.070-1.094 | 2.5 | 0.94 |
| 1017 | 3,4-dichlorocatechol | 1136 | 184 | 1.083-1.105 | 2.5 | 0.60 |
| 1119 | 4,5-dichlorocatechol- 13C6 | 1158 | 164 | 1.384-1.424 | ||
| 1219 | 4,5-dichlorocatechol | 1158 | 1119 | 0.998-1.001 | 2.5 | 0.24 |
| 1012 | 3,4,5-trichloroguaiacol | 1177 | 184 | 1.120-1.160 | 2.5 | 0.49 |
| 1114 | 4,5,6-trichloroguaiacol- 13C6 | 1208 | 164 | 1.444-1.484 | ||
| 1214 | 4,5,6-trichloroguaiacol | 1208 | 1114 | 0.998-1.002 | 2.5 | 0.25 |
| 1021 | 3,4,6-trichlorocatechol | 1213 | 184 | 1.155-1.185 | 5.0 | 0.44 |
| 1025 | 5,6-dichlorovanillin | 1246 | 184 | 1.182-1.222 | 5.0 | 0.80 |
| 1026 | 2-chlorosyringaldehyde | 1255 | 184 | 1.190-1.230 | 2.5 | 0.87 |
| 1107 | pentachlorophenol- 13C6 | 1267 | 164 | 1.511-1.561 | ||
| 1207 | pentachlorophenol | 1268 | 1107 | 0.998-1.002 | 5.0 | 0.28 |
| 1020 | 3,4,5-trichlorocatechol | 1268 | 184 | 1.208-1.238 | 5.0 | 0.53 |
| 1115 | tetrachloroguaiacol- 13C6 | 1289 | 164 | 1.537-1.587 | ||
| 1215 | tetrachloroguaiacol | 1290 | 1115 | 0.998-1.002 | 5.0 | 0.23 |
| 1028 | trichlorosyringol | 1301 | 184 | 1.240-1.270 | 2.5 | 0.64 |
| 1122 | tetrachlorocatechol- 13C6 | 1365 | 164 | 1.630-1.690 | ||
| 1222 | tetrachlorocatechol | 1365 | 1122 | 0.998-1.002 | 5.0 | 0.76 |
| 1027 | 2,6-dichlorosyringaldehyde | 1378 | 184 | 1.309-1.349 | 5.0 | 1.13 |
1 Four digit numbers beginning with 10 indicate a pollutant quantified by the internal standard method; four digit numbers beginning with 11 indicate a labeled compound quantified by the internal standard method; four digit numbers beginning with 12 indicate a pollutant quantified by isotope dilution.
2 The retention times in this column are based on data from a single laboratory (reference 12), utilizing the GC conditions in Section 11.
3 Relative retention time windows are estimated from EPA Method 1625.
4 The minimum level (ML) is defined as the level at which the entire analytical system must give a recognizable signal and acceptable calibration point for the analyte. It is equivalent to the concentration of the lowest calibration standard, assuming that all method-specified sample weights, volumes, and cleanup procedures have been employed.
5 40 CFR part 136, appendix B; from reference 2.
Table 3 - DFTPP Mass Intensity Specifications 1
| Mass | Intensity required |
|---|---|
| 51 | 8 to 82% of m/z 198. |
| 68 | Less than 2% of m/z 69. |
| 69 | 11 to 91% of m/z 198. |
| 70 | Less than 2% of m/z 69. |
| 127 | 32 to 59% of m/z 198. |
| 197 | Less than 1% of m/z 198. |
| 198 | Base peak, 100% abundance. |
| 199 | 4 to 9% of m/z 198. |
| 275 | 11 to 30% of m/z 198. |
| 441 | 44 to 110% of m/z 443. |
| 442 | 30 to 86% of m/z 198. |
| 443 | 14 to 24% of m/z 442. |
1 Reference 7.
Table 4 - Characteristic M/Z's of Chlorophenolic Compounds
| Compound | Primary m/z |
|---|---|
| 4-chlorophenol | 128 |
| 2,4-dichlorophenol | 162 |
| 2,4-dichlorophenol-d3 | 167 |
| 2,6-dichlorophenol | 162 |
| 2,4,5-trichlorophenol | 196 |
| 2,4,6-trichlorophenol | 196 |
| 2,3,4,6-tetrachlorophenol | 232 |
| pentachlorophenol | 266 |
| pentachlorophenol−13C6 | 272 |
| 4-chloroguaiacol | 158 |
| 4-chloroguaiacol−13C6 | 164 |
| 3,4-dichloroguaiacol | 192 |
| 4,5-dichloroguaiacol | 192 |
| 4,6-dichloroguaiacol | 192 |
| 3,4,5-trichloroguaiacol | 226 |
| 3,4,6-trichloroguaiacol | 226 |
| 4,5,6-trichloroguaiacol | 226 |
| 4,5,6-trichloroguaiacol−13C6 | 234 |
| tetrachloroguaiacol | 262 |
| tetrachloroguaiacol−13C6 | 268 |
| 4-chlorocatechol | 144 |
| 3,4-dichlorocatechol | 178 |
| 3,6-dichlorocatechol | 178 |
| 4,5-dichlorocatechol | 178 |
| 4,5-dichlorocatechol−13C6 | 184 |
| 3,4,5-trichlorocatechol | 212 |
| 3,4,6-trichlorocatechol | 212 |
| tetrachlorocatechol | 248 |
| tetrachlorocatechol−13C6 | 254 |
| 5-chlorovanillin | 186 |
| 5-chlorovanillin−13C6 | 192 |
| 6-chlorovanillin | 186 |
| 5,6-dichlorovanillin | 220 |
| 2-chlorosyringaldehyde | 216 |
| 2,6-dichlorosyringaldehyde | 250 |
| trichlorosyringol | 256 |
| Sample Matrix Internal Standard (SMIS) | |
| 3,4,5-trichlorophenol | 196 |
| Instrument Internal Standard (IIS) | |
| 2,2′-difluorobiphenyl | 190 |
Table 5 - Acceptance Criteria for Performance Tests 1
| EGD No. 2 | Compound | Test conc. 3 (µg/mL) | Initial precision and recovery sec. 9.3.2 (percent) | Ongoing recovery sec. 9.6 (percent) | Labeled compound and SMIS recovery sec. 9.4 and 14.6 | ||
|---|---|---|---|---|---|---|---|
| s | X | With ascorbic acid P (%) | Without ascorbic acid P (%) | ||||
| 1001 | 4-chlorophenol | 25 | 64 | 72-144 | 40-236 | ||
| 1202 | 2,4-dichlorophenol | 50 | 14 | 84-120 | 84-118 | ||
| 1102 | 2,4-dichlorophenol-d3 | 25 | 54 | 64-160 | 56-170 | 58-135 | 27-143 |
| 1003 | 2,6-dichlorophenol | 50 | 20 | 66-148 | 58-170 | ||
| 1004 | 2,4,5-trichlorophenol | 50 | 14 | 78-140 | 82-128 | ||
| 1005 | 2,4,6-trichlorophenol | 50 | 20 | 72-142 | 72-146 | ||
| 1006 | 2,3,4,6-tetrachlorophenol | 50 | 14 | 80-132 | 82-132 | ||
| 1207 | pentachlorophenol | 100 | 6 | 90-111 | 84-120 | ||
| 1107 | pentachlorophenol- 13C6 | 25 | 21 | 58-169 | 61-157 | 8-143 | 27-167 |
| 1208 | 4-chloroguaiacol | 25 | 20 | 88-120 | 88-120 | ||
| 1108 | 4-chloroguaiacol- 13C6 | 25 | 104 | 68-148 | 64-152 | 59-121 | 43-168 |
| 1009 | 3,4-dichloroguaiaco 4 | 50 | 18 | 80-126 | 82-126 | ||
| 1010 | 4,5-dichloroguaiacol | 50 | 14 | 82-121 | 80-128 | ||
| 1011 | 4,6-dichloroguaiacol | 50 | 16 | 82-126 | 86-120 | ||
| 1012 | 3,4,5-trichloroguaiacol | 50 | 16 | 78-130 | 80-134 | ||
| 1013 | 3,4,6-trichloroguaiacol | 50 | 16 | 64-152 | 74-140 | ||
| 1214 | 4,5,6-trichloroguaiacol | 50 | 14 | 92-106 | 88-116 | ||
| 1114 | 4,5,6-trichloroguaiacol- 13C6 | 25 | 48 | 66-146 | 74-140 | 48-131 | 51-139 |
| 1215 | tetrachloroguaiacol | 100 | 7 | 84-115 | 81-126 | ||
| 1115 | tetrachloroguaiacol- 13C6 | 25 | 22 | 57-173 | 65-161 | 35-120 | 27-161 |
| 1016 | 4-chlorocatechol | 25 | 48 | 76-140 | 80-124 | ||
| 1017 | 3,4-dichlorocatechol | 50 | 24 | 66-154 | 78-134 | ||
| 1018 | 3,6-dichlorocatechol | 50 | 16 | 78-136 | 84-126 | ||
| 1219 | 4,5-dichlorocatechol | 50 | 8 | 84-118 | 86-122 | ||
| 1119 | 4,5-dichlorocatechol- 13C6 | 25 | 78 | 68-144 | 66-142 | 33-129 | 0-190 |
| 1020 | 3,4,5-trichlorocatechol | 100 | 17 | 60-166 | 72-128 | ||
| 1021 | 3,4,6-trichlorocatechol 4 | 100 | 17 | 74-138 | 64-149 | ||
| 1222 | tetrachlorocatechol | 100 | 29 | 46-234 | 81-132 | ||
| 1122 | tetrachlorocatechol- 13C6 | 25 | 39 | 48-227 | 63-152 | 14-118 | 0-184 |
| 1223 | 5-chlorovanillin | 50 | 20 | 94-208 | 84-118 | ||
| 1123 | 5-chlorovanillin- 13C6 | 25 | 84 | 68-160 | 70-144 | 51-126 | 32-254 |
| 1024 | 6-chlorovanillin | 50 | 22 | 82-128 | 80-126 | ||
| 1025 | 5,6-dichlorovanillin | 100 | 9 | 67-146 | 77-140 | ||
| 1026 | 2-chlorosyringaldehyde | 50 | 28 | 76-130 | 72-156 | ||
| 1027 | 2,6-dichlorosyringaldehyde | 100 | 14 | 82-129 | 60-183 | ||
| 1028 | trichlorosyringol | 50 | 18 | 76-136 | 66-174 | ||
| Sample Matrix Internal Standard | |||||||
| 184 | 3,4,5-trichlorophenol | 100 | 47 | 62-185 | 68-144 | 56-116 | 24-167 |
1 Specifications derived from multi-laboratory testing of draft method.
2 Four-digit numbers beginning with 10 indicate a pollutant quantified by the internal standard method; four-digit numbers beginning with 11 indicate a labeled compound quantified by the internal standard method; four-digit numbers beginning with 12 indicate a pollutant quantified by isotope dilution.
3 Test concentrations are in units of µg/mL.
4 Specification derived from isomer.
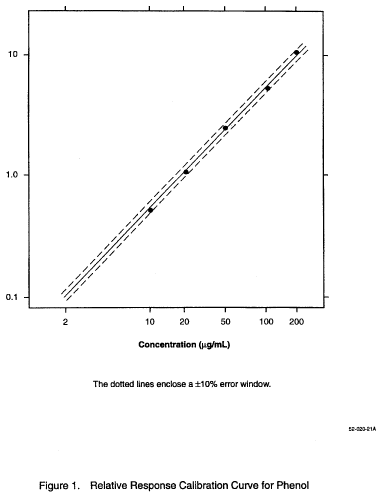
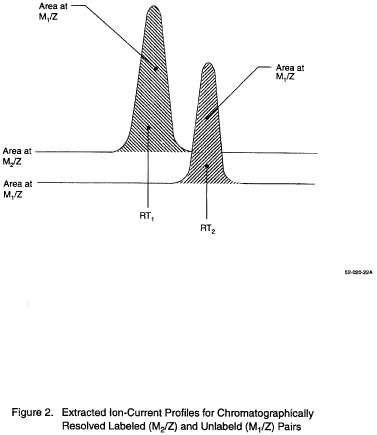
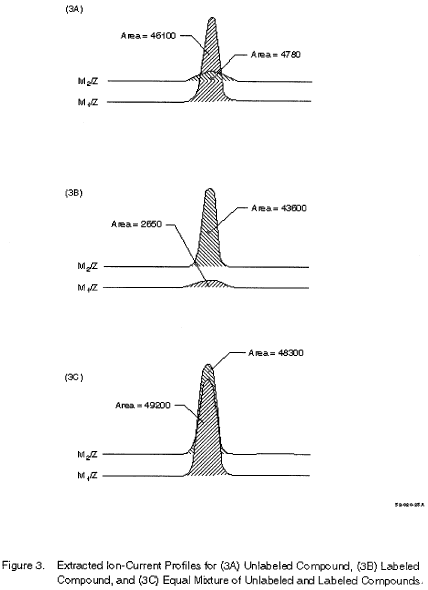
 20.0 Glossary of
Definitions and Purposes
20.0 Glossary of
Definitions and Purposes
These definitions and purposes are specific to this method but have been conformed to common usage as much as possible.
20.1 Units of weight and measure and their abbreviations
20.1.1 Symbols.
°C degrees Celsius µL microliter < less than > greater than % percent20.1.2 Alphabetical characters.
cm centimeter g gram h hour ID inside diameter in. inch L liter M Molecular ion m meter mg milligram min minute mL milliliter mm millimeter m/z mass-to-charge ratio N normal; gram molecular weight of solute divided by hydrogen equivalent of solute, per liter of solution OD outside diameter pg picogram ppb part-per-billion ppm part-per-million ppt part-per-trillion psig pounds-per-square inch gauge v/v volume per unit volume w/v weight per unit volume20.2 Definitions and acronyms (in alphabetical order).
Analyte: A chlorophenolic tested for by this method.
The analytes are listed in Table 1.
Calibration standard (CAL): A solution prepared from a secondary standard and/or stock solutions and used to calibrate the response of the instrument with respect to analyte concentration.
Calibration verification standard (VER): The mid-point calibration standard (CS3) that is used to verify calibration. See Table 4.
Chlorophenolics: collectively, the analytes listed in Table 1.
CS1, CS2, CS3, CS4, CS5: See Calibration standards and Table 4.
Field blank: An aliquot of reagent water or other reference matrix that is placed in a sample container in the laboratory or the field, and treated as a sample in all respects, including exposure to sampling site conditions, storage, preservation, and all analytical procedures. The purpose of the field blank is to determine if the field or sample transporting procedures and environments have contaminated the sample.
GC: Gas chromatograph or gas chromatography.
HRGC: High resolution GC.
IPR: Initial precision and recovery; four aliquots of the diluted PAR standard analyzed to establish the ability to generate acceptable precision and accuracy. An IPR is performed prior to the first time this method is used and any time the method or instrumentation is modified.
K-D: Kuderna-Danish concentrator; a device used to concentrate the analytes in a solvent.
Laboratory blank: See Method blank.
Laboratory control sample (LCS): See Ongoing precision and recovery standard (OPR).
Laboratory reagent blank: See Method blank.
May: This action, activity, or procedural step is neither required nor prohibited.
May not: This action, activity, or procedural step is prohibited.
Method blank: An aliquot of reagent water that is treated exactly as a sample including exposure to all glassware, equipment, solvents, reagents, internal standards, and surrogates that are used with samples. The method blank is used to determine if analytes or interferences are present in the laboratory environment, the reagents, or the apparatus.
Minimum level (ML): The level at which the entire analytical system must give a recognizable signal and acceptable calibration point for the analyte. It is equivalent to the concentration of the lowest calibration standard, assuming that all method-specified sample weights, volumes, and cleanup procedures have been employed.
MS: Mass spectrometer or mass spectrometry.
Must: This action, activity, or procedural step is required.
OPR: Ongoing precision and recovery standard (OPR); a laboratory blank spiked with known quantities of analytes. The OPR is analyzed exactly like a sample. Its purpose is to assure that the results produced by the laboratory remain within the limits specified in this method for precision and recovery.
PAR: Precision and recovery standard; secondary standard that is diluted and spiked to form the IPR and OPR.
Preparation blank: See Method blank.
Primary dilution standard: A solution containing the specified analytes that is purchased or prepared from stock solutions and diluted as needed to prepare calibration solutions and other solutions.
Quality control check sample (QCS): A sample containing all or a subset of the analytes at known concentrations. The QCS is obtained from a source external to the laboratory or is prepared from a source of standards different from the source of calibration standards. It is used to check laboratory performance with test materials prepared external to the normal preparation process.
Reagent water: Water demonstrated to be free from the analytes of interest and potentially interfering substances at the method detection limit for the analyte.
Relative standard deviation (RSD): The standard deviation times 100 divided by the mean.
RF: Response factor. See Section 10.5.1.
RR: Relative response. See Section 10.4.4.
RSD: See Relative standard deviation.
Should: This action, activity, or procedural step is suggested but not required.
Stock solution: A solution containing an analyte that is prepared using a reference material traceable to EPA, the National Institute of Science and Technology (NIST), or a source that will attest to the purity and authenticity of the reference material.
VER: See Calibration verification standard.